 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
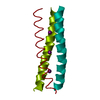
Yorodumi- PDB-2yo2: Salmonella enterica SadA 255-358 fused to GCN4 adaptors (SadAK12) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2yo2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Salmonella enterica SadA 255-358 fused to GCN4 adaptors (SadAK12) | ||||||
 Components Components | GENERAL CONTROL PROTEIN GCN4, PUTATIVE INNER MEMBRANE PROTEIN | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / FGG DOMAIN / DALL DOMAIN / DALL2 / TRIMERIC AUTOTRANSPORTER ADHESIN / TAA / CHIMERA | ||||||
| Function / homology |  Function and homology information Function and homology informationFCERI mediated MAPK activation / protein localization to nuclear periphery / Activation of the AP-1 family of transcription factors / negative regulation of ribosomal protein gene transcription by RNA polymerase II / positive regulation of cellular response to amino acid starvation / response to amino acid starvation / mediator complex binding / Oxidative Stress Induced Senescence / amino acid biosynthetic process / TFIID-class transcription factor complex binding ...FCERI mediated MAPK activation / protein localization to nuclear periphery / Activation of the AP-1 family of transcription factors / negative regulation of ribosomal protein gene transcription by RNA polymerase II / positive regulation of cellular response to amino acid starvation / response to amino acid starvation / mediator complex binding / Oxidative Stress Induced Senescence / amino acid biosynthetic process / TFIID-class transcription factor complex binding / positive regulation of transcription initiation by RNA polymerase II / positive regulation of RNA polymerase II transcription preinitiation complex assembly / cellular response to nutrient levels / cellular response to amino acid starvation / cell outer membrane / RNA polymerase II transcription regulator complex / protein transport / transcription regulator complex / DNA-binding transcription activator activity, RNA polymerase II-specific / sequence-specific DNA binding / RNA polymerase II-specific DNA-binding transcription factor binding / DNA-binding transcription factor activity, RNA polymerase II-specific / intracellular signal transduction / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / chromatin binding / negative regulation of transcription by RNA polymerase II / cell surface / positive regulation of transcription by RNA polymerase II / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |   SALMONELLA ENTERICA SUBSP. ENTERICA SEROVAR TYPHIMURIUM (bacteria) SALMONELLA ENTERICA SUBSP. ENTERICA SEROVAR TYPHIMURIUM (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Hartmann, M.D. / Hernandez Alvarez, B. / Lupas, A.N. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2012 Title: Complete Fiber Structures of Complex Trimeric Autotransporter Adhesins Conserved in Enterobacteria. Authors: Hartmann, M.D. / Grin, I. / Dunin-Horkawicz, S. / Deiss, S. / Linke, D. / Lupas, A.N. / Hernandez Alvarez, B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2yo2.cif.gz | 80.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2yo2.ent.gz | 61.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2yo2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yo/2yo2ftp://data.pdbj.org/pub/pdb/validation_reports/yo/2yo2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2ynyC  2ynzC  2yo0C  2yo1C  2yo3C  1gcmS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








































































-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 18397.605 Da / Num. of mol.: 1 Fragment: RESIDUES 255-358 FUSED TO GCN4 ADAPTORS AT EITHER END, RESIDUES 250-278 Mutation: YES Source method: isolated from a genetically manipulated source Details: N- AND C-TERMINAL IN-REGISTER FUSION TO GCN4 ADAPTORS Source: (gene. exp.) SALMONELLA ENTERICA SUBSP. ENTERICA SEROVAR TYPHIMURIUM (bacteria)Production host: |
|---|---|
| #2: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 101 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 101 / Source method: isolated from a natural source / Formula: H2O |
| Sequence details | C-TERMINAL HIS-TAG |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.3 Å3/Da / Density % sol: 62 % / Description: NONE |
|---|---|
| Crystal grow | Details: 500 MM AMMONIUM TARTRATE, 100 MM SODIUM ACETATE PH 5.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 1 / Beamline: X10SA / Wavelength: 1 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: May 19, 2009 |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2→38.3 Å / Num. obs: 18662 / % possible obs: 99.5 % / Redundancy: 7.3 % / Rmerge(I) obs: 0.09 / Net I/σ(I): 13.8 |
| Reflection shell | Resolution: 2→2.12 Å / Redundancy: 6.01 % / Rmerge(I) obs: 0.64 / Mean I/σ(I) obs: 2.29 / % possible all: 98.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1GCM Resolution: 2→38.26 Å / Cor.coef. Fo:Fc: 0.914 / Cor.coef. Fo:Fc free: 0.902 / SU B: 8.515 / SU ML: 0.108 / Cross valid method: THROUGHOUT / ESU R: 0.158 / ESU R Free: 0.156 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES WITH TLS ADDED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.146 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→38.26 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|