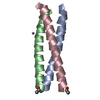
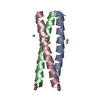
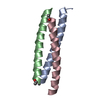
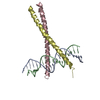
Journal: Biochemistry / Year: 2006 Title: Coiled Coils at the Edge of Configurational Heterogeneity. Structural Analyses of Parallel and Antiparallel Homotetrameric Coiled Coils Reveal Configurational Sensitivity to a Single Solvent- ...Title: Coiled Coils at the Edge of Configurational Heterogeneity. Structural Analyses of Parallel and Antiparallel Homotetrameric Coiled Coils Reveal Configurational Sensitivity to a Single Solvent-Exposed Amino Acid Substitution. Authors: Yadav, M.K. / Leman, L.J. / Price, D.J. / Brooks, C.L. / Stout, C.D. / Ghadiri, M.R.
Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O
Compound details
ENGINEERED MUTATION IN CHAINS A, B, C, D: ARG 251 TO LYS ENGINEERED MUTATION IN CHAINS A, B, C, D: ...ENGINEERED MUTATION IN CHAINS A, B, C, D: ARG 251 TO LYS ENGINEERED MUTATION IN CHAINS A, B, C, D: ARG 256 TO LYS ENGINEERED MUTATION IN CHAINS A, B, C, D: CYS 268 TO GLU ENGINEERED MUTATION IN CHAINS A, B, C, D: ARG 275 TO LYS ENGINEERED MUTATION IN CHAINS A, B, C, D: ARG 276 TO LYS ENGINEERED MUTATION IN CHAINS A, B, C, D: TYR 265 TO HIS
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 3
-
Sample preparation
Crystal
Density Matthews: 2 Å3/Da / Density % sol: 38.9 % Description: STRUCTURE WAS ORIGINALLY SOLVED BETWEEN SIRAS PHASING BETWEEN 2SEMET PEPTIDE AND IT'S DERIVATIVE SOAKED WITH 50MM KAUCN2
Crystal grow
Method: vapor diffusion, hanging drop / pH: 5 Details: HANGING DROP, 1UL OF 10MG/ML PEPTIDE IN WATER, 1UL 100MM CAPS, 30% PEG 400, PH 10.5, 1UL OF 50MM TCEP IN WATER
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads




























































 PDBj
PDBj

 Assembly
Assembly


 Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing