 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1swi | ||||||
|---|---|---|---|---|---|---|---|
| Title | GCN4-LEUCINE ZIPPER CORE MUTANT AS N16A COMPLEXED WITH BENZENE | ||||||
 Components Components | GCN4P1 | ||||||
 Keywords Keywords | LEUCINE ZIPPER / COILED COIL | ||||||
| Function / homology |  Function and homology information Function and homology informationFCERI mediated MAPK activation / protein localization to nuclear periphery / Activation of the AP-1 family of transcription factors / negative regulation of ribosomal protein gene transcription by RNA polymerase II / positive regulation of cellular response to amino acid starvation / response to amino acid starvation / mediator complex binding / Oxidative Stress Induced Senescence / amino acid biosynthetic process / TFIID-class transcription factor complex binding ...FCERI mediated MAPK activation / protein localization to nuclear periphery / Activation of the AP-1 family of transcription factors / negative regulation of ribosomal protein gene transcription by RNA polymerase II / positive regulation of cellular response to amino acid starvation / response to amino acid starvation / mediator complex binding / Oxidative Stress Induced Senescence / amino acid biosynthetic process / TFIID-class transcription factor complex binding / positive regulation of RNA polymerase II transcription preinitiation complex assembly / positive regulation of transcription initiation by RNA polymerase II / cellular response to nutrient levels / cellular response to amino acid starvation / RNA polymerase II transcription regulator complex / DNA-binding transcription activator activity, RNA polymerase II-specific / transcription regulator complex / sequence-specific DNA binding / RNA polymerase II-specific DNA-binding transcription factor binding / DNA-binding transcription factor activity, RNA polymerase II-specific / intracellular signal transduction / RNA polymerase II cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / chromatin binding / negative regulation of transcription by RNA polymerase II / positive regulation of transcription by RNA polymerase II / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.6 Å X-RAY DIFFRACTION / Resolution: 2.6 Å | ||||||
 Authors Authors | Gonzalez, L. / Plecs, J. / Alber, T. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1996 Title: An engineered allosteric switch in leucine-zipper oligomerization. Authors: Gonzalez Jr., L. / Plecs, J.J. / Alber, T. #1: Journal: Nature / Year: 1994Title: Crystal Structure of an Isoleucine-Zipper Trimer Authors: Harbury, P.B. / Kim, P.S. / Alber, T. #2: Journal: Science / Year: 1993Title: A Switch between Two-, Three-, and Four-Stranded Coiled Coils in GCN4 Leucine Zipper Mutants Authors: Harbury, P.B. / Zhang, T. / Kim, P.S. / Alber, T. #3: Journal: Science / Year: 1991Title: X-Ray Structure of the GCN4 Leucine Zipper Authors: O'Shea, E.K. / Klemm, J.D. / Kim, P.S. / Alber, T. | ||||||
| History |
|
- Structure visualization
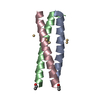
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1swi.cif.gz | 28.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1swi.ent.gz | 19.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1swi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sw/1swiftp://data.pdbj.org/pub/pdb/validation_reports/sw/1swi | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein/peptide | Mass: 3962.639 Da / Num. of mol.: 3 / Mutation: N16A Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P03069 #2: Chemical | ChemComp-BNZ / | 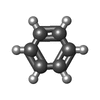  Mass: 78.112 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6 Mass: 78.112 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H6#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 29 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.88 Å3/Da / Density % sol: 34 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS pH: 7 / Method: vapor diffusion | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction source | Wavelength: 1.5418 |
|---|---|
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Aug 1, 1994 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Num. obs: 2618 / % possible obs: 86 % / Observed criterion σ(I): 2 / Redundancy: 2.3 % / Rmerge(I) obs: 0.097 |
| Reflection | *PLUS Highest resolution: 2.6 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.6→6 Å / σ(F): 2 Details: ELECTRON DENSITY FOR THE N-TERMINAL HEPTAD (RESIDUES 1 - 7) IS COMPARATIVELY WEAK, AND THE MAIN CHAIN B VALUES ARE ABOVE AVERAGE. THE N-TERMINAL HEPTAD MAKES RELATIVELY FEW CRYSTAL CONTACTS ...Details: ELECTRON DENSITY FOR THE N-TERMINAL HEPTAD (RESIDUES 1 - 7) IS COMPARATIVELY WEAK, AND THE MAIN CHAIN B VALUES ARE ABOVE AVERAGE. THE N-TERMINAL HEPTAD MAKES RELATIVELY FEW CRYSTAL CONTACTS AND MAY BE DYNAMICALLY DISORDERED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 44 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
