 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h87: Gadolinium derivative of tetragonal Hen Egg-White Lysozyme at 1.7... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h87 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Gadolinium derivative of tetragonal Hen Egg-White Lysozyme at 1.7 A resolution | ||||||
 Components Components | LYSOZYME C | ||||||
 Keywords Keywords | HYDROLASE / GADOLINIUM DERIVATIVE / LYSOZYME / O-GLYCOSYL HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.72 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.72 Å | ||||||
 Authors Authors | Girard, E. / Chantalat, L. / Vicat, J. / Kahn, R. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2001 Title: Gd-Hp-Do3A, a Complex to Obtain High-Phasing-Power Heavy Atom Derivatives for Sad and MAD Experiments. Results with Tetragonal Hen Egg-White Lysozyme Authors: Girard, E. / Chantalat, L. / Vicat, J. / Kahn, R. #1: Journal: J.Mol.Biol. / Year: 1994 Title: Thermal Expansion of Hen-Egg-White Lysozyme. Comparison of the 1.9 Angstroms Resolution Structures of the Tetragonal Form of the Enzyme at 100K and 298K Authors: Young, A.C.M. / Tilton, R.F. / Dewan, J.C. #2: Journal: J.Synchrotron Radia. / Year: 2001 Title: High-Pressure Protein Crystallography (Hppx): Instrumentation, Methodology and Results on Lysozyme Crystals Authors: Fourme, R. / Kahn, R. / Mezouar, M. / Girard, E. / Hoerentrup, C. / Prange, T. / Ascone, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h87.cif.gz | 47.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h87.ent.gz | 33.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1h87.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h8/1h87ftp://data.pdbj.org/pub/pdb/validation_reports/h8/1h87 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6lytS S: Starting model for refinement |
|---|---|
| Similar structure data |

































































































































-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: PURCHASED FROM BOEHRINGER MANNHEIM BATCH NUMBER 13032022-90 Source: (natural) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | 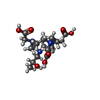  Mass: 404.459 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H32N4O7 Mass: 404.459 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C17H32N4O7#3: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#4: Chemical |   Mass: 157.250 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Gd Mass: 157.250 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Gd#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 154 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 154 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.02 Å3/Da / Density % sol: 38.55 % | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / pH: 4.5 Details: NACL 0.8 M, SODIUM ACETATE 50 MM, PH4.5, GD-HP-DO3A 100MM, [PROT]=40 MG/ML, 293 K, pH 4.50 | ||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jan 15, 2000 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.72→17.2 Å / Num. obs: 12876 / % possible obs: 99.5 % / Observed criterion σ(I): 3 / Redundancy: 12.5 % / Biso Wilson estimate: 15.5 Å2 / Rsym value: 0.059 / Net I/σ(I): 8.1 |
| Reflection shell | Resolution: 1.72→1.81 Å / Redundancy: 8.9 % / Mean I/σ(I) obs: 4 / Rsym value: 0.173 / % possible all: 96.8 |
| Reflection | *PLUS Num. measured all: 166568 / Rmerge(I) obs: 0.059 |
| Reflection shell | *PLUS % possible obs: 96.8 % / Rmerge(I) obs: 0.173 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PBD ENTRY 6LYT Resolution: 1.72→27.33 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 1120223.55 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: SOME OF THE SIDE CHAIN ATOMS OF RESIDUES ARG21, ARG45, ARG61 AND ARG73 HAVE A POORLY DEFINED DENSITY.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 43.6549 Å2 / ksol: 0.336137 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.72→27.33 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.72→1.83 Å / Rfactor Rfree error: 0.017 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.18 / Rfactor Rfree: 0.21 / Rfactor Rwork: 0.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.25 |
