 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lkr | ||||||
|---|---|---|---|---|---|---|---|
| Title | MONOCLINIC HEN EGG WHITE LYSOZYME IODIDE | ||||||
 Components Components | LYSOZYME | ||||||
 Keywords Keywords | HYDROLASE / GLYCOSIDASE / LYSOZYME | ||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Steinrauf, L.K. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 1998 Title: Structures of monoclinic lysozyme iodide at 1.6 A and of triclinic lysozyme nitrate at 1.1 A. Authors: Steinrauf, L.K. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1996Title: Studies of Monoclinic Hen Egg-White Lysozyme. Iv. X-Ray Refinement at 1.8 A Resolution and a Comparison of the Variable Regions in the Polymorphic Forms Authors: Rao, S.T. / Sundaralingam, M. #2: Journal: Acta Crystallogr.,Sect.C / Year: 1983Title: Studies of Monoclinic Hen Egg White Lysozyme. II. The Refinement at 2.5 A Resolution--Conformational Variability between the Two Independent Molecules Authors: Rao, S.T. / Hogle, J. / Sundaralingam, M. #3: Journal: Acta Crystallogr. / Year: 1959Title: Preliminary X-Ray Data for Some New Crystalline Forms of Beta-Lactoglobulin and Hen Egg-White Lysozyme Authors: Steinrauf, L.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lkr.cif.gz | 145.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lkr.ent.gz | 99.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1lkr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lk/1lkrftp://data.pdbj.org/pub/pdb/validation_reports/lk/1lkr | HTTPS FTP |
|---|
-Related structure data















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 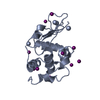
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.975515, 0.165171, -0.14522), Vector: |
-Components
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 2 / Source method: isolated from a natural source Details: WORTHINGTON BIOCHEMICALS, USED WITHOUT PURIFICATION Source: (natural) #2: Chemical | ChemComp-IOD /   Mass: 126.904 Da / Num. of mol.: 17 / Source method: obtained synthetically / Formula: I Mass: 126.904 Da / Num. of mol.: 17 / Source method: obtained synthetically / Formula: I#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 311 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 311 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.85 Å3/Da / Density % sol: 26 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8 Details: 1% PROTEIN, 200 MMOLAR ACETATE BUFFER, PH 8, 5% SODIUM IODIDE, pH 8.0 | |||||||||||||||||||||||||
| Crystal grow | *PLUS Method: batch method | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Sep 1, 1994 |
| Radiation | Monochromator: BENT CYLINDRICAL GE(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→8 Å / Num. obs: 22327 / % possible obs: 63 % / Observed criterion σ(I): 1 / Redundancy: 3 % / Rmerge(I) obs: 0.0761 |
| Reflection | *PLUS Num. measured all: 68962 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.6→8 Å / Cross valid method: FREE R / σ(F): 1 / Stereochemistry target values: ENGH AND HUBER Details: REFINEMENT WAS INITIATED BY PROLSQ, WHICH DID NOT ALLOW ANISOTROPY. THE FINAL REFINEMENT WAS CARRIED OUT USING SHELX-L93 WITH ALL ATOMS ANISOTROPIC. THE PRESENCE OF THE 17 IODIDE ATOMS MAKES ...Details: REFINEMENT WAS INITIATED BY PROLSQ, WHICH DID NOT ALLOW ANISOTROPY. THE FINAL REFINEMENT WAS CARRIED OUT USING SHELX-L93 WITH ALL ATOMS ANISOTROPIC. THE PRESENCE OF THE 17 IODIDE ATOMS MAKES THE APPLICATION OF THE USUAL ANALYSIS OF ERRORS NOT VALID. ONLY WHEN ANOTHER MONOCLINIC LYSOZYME STRUCTURE WITHOUT HEAVY ATOMS HAS BEEN REFINED BY THE SAME PROCEDURE WILL AN ANALYSIS BE POSSIBLE. NEVERTHELESS, A COMPARISON OF THE BOND LENGTHS BETWEEN THE TWO MOLECULES OF LYSOZYME GIVES AN AVERAGE ERROR OF 0.12 ANGSTROMS, WHICH IS IN GOOD AGREEMENT WITH THE LUZATTI ANALYSIS.
| |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→8 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
