Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 1.84 Å3/Da / Density % sol: 33.2 %
Crystal grow
Temperature: 294 K / Method: batch mode / pH: 4.7 Details: CRYSTALLIZATION CONDITIONS: 40MG HEWL CO-CRYSTALLISED WITH 2.6MG CISPLATIN, WITH THE PLATINUM COMPOUNDS BEING IN A 3-FOLD MOLAR EXCESS TO THE PROTEIN, 462.5 UL OF A 0.02M NAAC SOLUTION ALONG ...Details: CRYSTALLIZATION CONDITIONS: 40MG HEWL CO-CRYSTALLISED WITH 2.6MG CISPLATIN, WITH THE PLATINUM COMPOUNDS BEING IN A 3-FOLD MOLAR EXCESS TO THE PROTEIN, 462.5 UL OF A 0.02M NAAC SOLUTION ALONG WITH 462.5 UL OF A 0.5M NANO3 SOLUTION WAS USED WITH 75 UL DMSO ADDED, BATCH, PH 4.7, EVAPORATION, TEMPERATURE 294K
-
Data collection
Diffraction
Mean temperature: 294 K
Diffraction source
Source: ROTATING ANODE / Type: BRUKER AXS MICROSTAR-H / Wavelength: 1.5418 Å
Detector
Type: APEX II CCD / Detector: CCD / Date: Dec 15, 2012
Radiation
Monochromator: CONFOCAL MIRROR OPTICS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.5418 Å / Relative weight: 1
Reflection
Resolution: 1.42→32.31 Å / Num. obs: 18176 / % possible obs: 94.8 % / Redundancy: 5.7 % / Rmerge(I) obs: 0.145 / Net I/σ(I): 6.8
Reflection shell
Resolution: 1.42→1.51 Å / Redundancy: 2 % / Rmerge(I) obs: 0.554 / Mean I/σ(I) obs: 1.1 / % possible all: 98.5
-
Processing
Software
Name
Version
Classification
REFMAC
5.7.0032
refinement
SAINT
datareduction
APEX
datascaling
PHASER
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4mwn
Resolution: 1.42→32.31 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.935 / SU B: 3.252 / SU ML: 0.058 / SU R Cruickshank DPI: 0.096 / Cross valid method: THROUGHOUT / ESU R: 0.093 / ESU R Free: 0.083 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.23687
984
5.1 %
RANDOM
Rwork
0.18462
-
-
-
obs
0.18734
18176
99.21 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United Kingdom, 1items
United Kingdom, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads





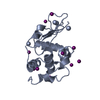







 PDBj
PDBj





 Assembly
Assembly

 Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM
Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM
 Mass: 62.005 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: NO3
Mass: 62.005 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: NO3
 Mass: 195.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Pt
Mass: 195.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Pt Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 37 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing