 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lcn | ||||||
|---|---|---|---|---|---|---|---|
| Title | Monoclinic hen egg white lysozyme, thiocyanate complex | ||||||
 Components Components | PROTEIN (LYSOZYME) | ||||||
 Keywords Keywords | HYDROLASE / GLYCOSIDASE / LYSOZYME | ||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.63 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.63 Å | ||||||
 Authors Authors | Hamiaux, C. / Prange, T. / Ducruix, A. / Vaney, M.C. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2001 Title: Structural effects of monovalent anions on polymorphic lysozyme crystals. Authors: Vaney, M.C. / Broutin, I. / Retailleau, P. / Douangamath, A. / Lafont, S. / Hamiaux, C. / Prange, T. / Ducruix, A. / Ries-Kautt, M. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1999Title: The Decameric Structure of Bovine Pancreatic Trypsin Inhibitor (BPTI) at 2.7 A Resolution. Authors: Hamiaux, C. / Prange, T. / Ries-Kautt, M. / Ducruix, A. / Lafont, S. / Astier, J.P. / Veesler, S. #2: Journal: Acta Crystallogr.,Sect.D / Year: 1998Title: Structures of Monoclinic Lysozyme Iodide at 1.6 A and of Triclinic Lysozyme Nitrate at 1.1 A Authors: Steinhauf, L.K. #3: Journal: Biophys.Chem. / Year: 1998Title: Characterization of the Interaction Between Bovine Pancreatic Trypsin Inhibitor and Thiocyanate by NMR Authors: Jolivalt, C. / Bockmann, A. / Ries-Kautt, M. / Ducruix, A. / Guittet, E. #4: Journal: Acta Crystallogr.,Sect.D / Year: 1995Title: Structure of Hexagonal Turkey Egg-White Lysozyme at 1.65 A Resolution Authors: Howell, P.L. #5: Journal: Acta Crystallogr.,Sect.B / Year: 1992Title: Structure Determination of a Dimeric Form of Erabutoxin-B, Crystallized from a Thiocyanate Solution Authors: Saludjian, P. / Prange, T. / Navaza, J. / Menez, R. / Guilloteau, J.P. / Ries-Kautt, M. / Ducruix, A. #6: Journal: J.Cryst.Growth / Year: 1991Title: Crystallisation of Basic Proteins by Ion Pairing Authors: Ries-Kautt, M. / Ducruix, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lcn.cif.gz | 64.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lcn.ent.gz | 47.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1lcn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lc/1lcnftp://data.pdbj.org/pub/pdb/validation_reports/lc/1lcn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1b0dC  1b2kC  1hf4C  5lymS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.97654, 0.1823, -0.11462), Vector: |
-Components
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 2 / Source method: isolated from a natural source Details: SIGMA, USED AFTER ION-EXCHANGE PURIFICATION (DESALTING) Source: (natural) #2: Chemical | 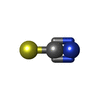  Mass: 58.082 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: CNS Mass: 58.082 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: CNS#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 136 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 136 / Source method: isolated from a natural source / Formula: H2OCompound details | LOOP 67-73 STANDS IN TWO DIFFERENT CONFORMATIONS IN MOLECULES A AND B, AND IS BETTER DEFINED IN ...LOOP 67-73 STANDS IN TWO DIFFERENT CONFORMATI | Has protein modification | Y | Nonpolymer details | 3 THIOCYANATE ANIONS ARE FOUND IN THE STRUCTURE. THEY ARE SPREAD OVER TWO SITES: SCN201 IS BOUND TO ...3 THIOCYANAT | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.9 Å3/Da / Density % sol: 33 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5 / Details: KSCN 190MM BUFFER CH3COOK 50MM, PH=5, pH 5.0 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 291 K / pH: 4.5 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 283 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LURE  / Beamline: DW32 / Wavelength: 0.963 / Beamline: DW32 / Wavelength: 0.963 |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Sep 15, 1998 / Details: MIRROR |
| Radiation | Monochromator: BENT CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.963 Å / Relative weight: 1 |
| Reflection | Resolution: 1.63→15.83 Å / Num. obs: 25539 / % possible obs: 96.8 % / Observed criterion σ(I): 1 / Redundancy: 4.2 % / Biso Wilson estimate: 18.4 Å2 / Rsym value: 0.028 / Net I/σ(I): 15.2 |
| Reflection shell | Resolution: 1.63→1.69 Å / Redundancy: 3.9 % / Mean I/σ(I) obs: 6.3 / Rsym value: 0.121 / % possible all: 89.5 |
| Reflection | *PLUS Rmerge(I) obs: 0.028 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: MONOMER A FROM 5LYM PDB ENTRY (MONOCLINIC LYSOZYME) Resolution: 1.63→15.83 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: FREE R / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.2 Å / Luzzati d res low obs: 5 Å / Luzzati sigma a obs: 0.19 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.63→15.83 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.63→1.69 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 10
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 2 / % reflection Rfree: 9.9 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 16.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.304 / % reflection Rfree: 9.9 % / Rfactor Rwork: 0.284 |
