 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1b0d: Structural effects of monovalent anions on polymorphic lysozyme c... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1b0d | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural effects of monovalent anions on polymorphic lysozyme crystals | ||||||
 Components Components | LYSOZYME | ||||||
 Keywords Keywords | HYDROLASE / O-GLYCOSYL | ||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 1.84 Å X-RAY DIFFRACTION / OTHER / Resolution: 1.84 Å | ||||||
 Authors Authors | Vaney, M.C. / Broutin, I. / Retailleau, P. / Lafont, S. / Hamiaux, C. / Prange, T. / Ries-Kautt, M. / Ducruix, A. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2001 Title: Structural effects of monovalent anions on polymorphic lysozyme crystals. Authors: Vaney, M.C. / Broutin, I. / Retailleau, P. / Douangamath, A. / Lafont, S. / Hamiaux, C. / Prange, T. / Ducruix, A. / Ries-Kautt, M. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1998Title: Refinement of Triclinic Hen Egg-White Lysozyme at Atomic Resolution Authors: Walsh, M.A. / Schneider, T.R. / Sieker, L.C. / Dauter, Z. / Lamzin, V.S. / Wilson, K.S. #2: Journal: Acta Crystallogr.,Sect.D / Year: 1998Title: Structures of Monoclinic Lysozyme Iodide at 1.6 Ang. and of Triclinic Lysozyme Nitrate at 1.1 Ang Authors: Steinrauf, L.K. #3: Journal: Acta Crystallogr.,Sect.D / Year: 1998Title: Locations of Bromide Ions in Tetragonal Lysozyme Crystals Authors: Lim, K. / Nadarajah, A. / Forsythe, E.L. / Pusey, M.L. #4: Journal: Acta Crystallogr.,Sect.D / Year: 1996Title: High-Resolution Structure (1.33 Ang.) Of a Hewl Lysozyme Tetragonal Crystal Grown in the Apcf Apparatus. Data and Structural Comparison with a Crystal Grown Under Microgravity from Spachab-01 Mission Authors: Vaney, M.C. / Maignan, S. / Ries-Kautt, M. / Ducruix, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1b0d.cif.gz | 39.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1b0d.ent.gz | 27.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1b0d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b0/1b0dftp://data.pdbj.org/pub/pdb/validation_reports/b0/1b0d | HTTPS FTP |
|---|
-Related structure data
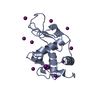
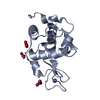
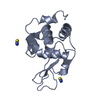




















-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) |
|---|---|
| #2: Chemical | ChemComp-TSU / 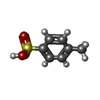  Mass: 172.202 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8O3S Mass: 172.202 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C7H8O3S |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.07 Å3/Da / Density % sol: 40.58 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, hanging drop / pH: 4.5 Details: THE CRYSTAL WAS OBTAINED IN PRESENCE OF NAPTS WITH A FINAL PROTEIN CONCENTRATION OF 40.5 MG/ML IN HANGING DROPS USING VAPOR DIFFUSION METHOD. THE EQUILIBRATION AT 291K WAS DONE AGAINST A ...Details: THE CRYSTAL WAS OBTAINED IN PRESENCE OF NAPTS WITH A FINAL PROTEIN CONCENTRATION OF 40.5 MG/ML IN HANGING DROPS USING VAPOR DIFFUSION METHOD. THE EQUILIBRATION AT 291K WAS DONE AGAINST A RESERVOIR SOLUTION CONTAINING 90 MM NAPTS, 50 MM SODIUM ACETATE ADJUSTED AT PH 4.5., vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 291 K / Method: vapor diffusion, hanging dropDetails: drop consists of equal volume of protein and reservoir solutions | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Apr 1, 1994 |
| Radiation | Protocol: MONOCHROMATIC / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.85→13.5 Å / Num. obs: 10462 / % possible obs: 97.5 % / Observed criterion σ(I): 0 / Redundancy: 6.6 % / Biso Wilson estimate: 21.4 Å2 / Rsym value: 0.049 / Net I/σ(I): 10.5 |
| Reflection shell | Resolution: 1.85→1.9 Å / Redundancy: 4.1 % / Mean I/σ(I) obs: 4.5 / Rsym value: 0.135 / % possible all: 85.8 |
| Reflection | *PLUS Num. obs: 10484 / Rmerge(I) obs: 0.049 |
| Reflection shell | *PLUS % possible obs: 85.8 % / Rmerge(I) obs: 0.135 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 1.84→14 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: SOME OF THE SIDE CHAIN ATOMS OF RESIDUES ARG 61, ARG 73, ASN 77, LYS 97, ASP 101, ASN 103, GLN 121 AND ARG 125 HAVE A POORLY DEFINED DENSITY. THESE ATOMS WERE BUILT USING STEREOCHEMICAL ...Details: SOME OF THE SIDE CHAIN ATOMS OF RESIDUES ARG 61, ARG 73, ASN 77, LYS 97, ASP 101, ASN 103, GLN 121 AND ARG 125 HAVE A POORLY DEFINED DENSITY. THESE ATOMS WERE BUILT USING STEREOCHEMICAL CONSTRAINTS AND KEPT IN THE COORDINATE FILE.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.84→14 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.84→1.92 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
