 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hf4: STRUCTURAL EFFECTS OF MONOVALENT ANIONS ON POLYMORPHIC LYSOZYME C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hf4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURAL EFFECTS OF MONOVALENT ANIONS ON POLYMORPHIC LYSOZYME CRYSTALS | ||||||
 Components Components | LYSOZYME | ||||||
 Keywords Keywords | HYDROLASE / HYDROLASE (O-GLYCOSYL) | ||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | ||||||
 Authors Authors | Vaney, M.C. / Broutin, I. / Ries-Kautt, M. / Ducruix, A. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2001 Title: Structural Effects of Monovalent Anions on Polymorphic Lysozyme Crystals Authors: Vaney, M.C. / Broutin, I. / Retailleau, P. / Douangamath, A. / Lafont, S. / Hamiaux, C. / Prange, T. / Ducruix, A. / Ries-Kautt, M. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Novel Approach to Phasing Proteins: Derivatization by Short Cryo-Soaking with Halides Authors: Dauter, Z. / Dauter, M. / Rajashankar, K.R. #2: Journal: J.Mol.Biol. / Year: 1999Title: Anomalous Signal of Solvent Bromides Used for Phasing of Lysozyme Authors: Dauter, Z. / Dauter, M. #3: Journal: Acta Crystallogr.,Sect.D / Year: 1998Title: Refinement of Triclinic Hen Egg-White Lysozyme at Atomic Resolution Authors: Walsh, M.A. / Schneider, T.R. / Sieker, L.C. / Dauter, Z. / Lamzin, V.S. / Wilson, K.S. #4: Journal: Acta Crystallogr.,Sect.D / Year: 1998Title: Structures of Monoclinic Lysozyme Iodide at 1.6 A and of Triclinic Lysozyme Nitrate at 1.1 A. Authors: Steinrauf, L.K. #5: Journal: Acta Crystallogr.,Sect.D / Year: 1998Title: Locations of Bromide Ions in Tetragonal Lysozyme Crystals Authors: Lim, K. / Nadarajah, A. / Forsythe, E.L. / Pusey, M.L. #6: Journal: Acta Crystallogr.,Sect.D / Year: 1996Title: High-Resolution Structure (1.33 A) of a Hew Lysozyme Tetragonal Crystal Grown in the Apcf Apparatus. Data and Structural Comparison with a Crystal Grown Under Microgravity from Spacehab-01 Mission. Authors: Vaney, M.C. / Maignan, S. / Ries-Kautt, M. / Ducruix, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hf4.cif.gz | 72.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hf4.ent.gz | 53.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1hf4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hf/1hf4ftp://data.pdbj.org/pub/pdb/validation_reports/hf/1hf4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1b0dC  1b2kC  1lcnC  5lymS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |
















































































-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 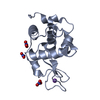
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | BIOLOGICAL_UNIT: MONOMER |
-Components
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) #2: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#3: Chemical | ChemComp-NO3 /   Mass: 62.005 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: NO3 Mass: 62.005 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: NO3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 251 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 251 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.63 Å3/Da / Density % sol: 24 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / pH: 4.5 Details: CRYSTALS OF NITRATE-LYSOZYME WERE GROWN UNDER A MICRO GRAVITY ENVIRONMENT DURING THE USML-2 MISSION LAUNCHED ON OCTOBER 1995 (SHUTTLE COLUMBIA). THE CRYSTALLIZATION SETUP WAS THE APCF ...Details: CRYSTALS OF NITRATE-LYSOZYME WERE GROWN UNDER A MICRO GRAVITY ENVIRONMENT DURING THE USML-2 MISSION LAUNCHED ON OCTOBER 1995 (SHUTTLE COLUMBIA). THE CRYSTALLIZATION SETUP WAS THE APCF DEVELOPPED BY EUROPEAN SPACE AGENCY. PROTEIN CONCENTRATION 10 MG/ML IN 50 MM SODIUM ACETATE BUFFER AT PH 4.5. GRADIENT CONCENTRATION APPLIED FROM 0 TO FINAL 290 MM AT 293K | ||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 293 K / pH: 5 / Method: microdialysis | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: LURE  / Beamline: DW32 / Wavelength: 0.97 / Beamline: DW32 / Wavelength: 0.97 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jan 15, 1996 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→10 Å / Num. obs: 35206 / % possible obs: 95.5 % / Observed criterion σ(I): 0 / Redundancy: 7.7 % / Biso Wilson estimate: 13.7 Å2 / Rmerge(I) obs: 0.12 / Net I/σ(I): 3.1 |
| Reflection shell | Resolution: 1.45→1.49 Å / Redundancy: 3.6 % / Rmerge(I) obs: 0.16 / Mean I/σ(I) obs: 4.5 / % possible all: 87 |
| Reflection | *PLUS Rmerge(I) obs: 0.123 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5LYM Resolution: 1.45→6 Å / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: THE LOOP ARG73-ASN74 OF CHAIN B WAS MODELED WITH ALTERNATE POSITIONS. IN CONTACT WITH THESE TWO LOOPS CONFORMATIONS, TWO SODIUM IONS NA+ WERE REFINED WITH ALTERNATE POSITIONS. ATOM ...Details: THE LOOP ARG73-ASN74 OF CHAIN B WAS MODELED WITH ALTERNATE POSITIONS. IN CONTACT WITH THESE TWO LOOPS CONFORMATIONS, TWO SODIUM IONS NA+ WERE REFINED WITH ALTERNATE POSITIONS. ATOM OCCUPANCIES WERE REFINED FOR NA+ 350 AND SOME NITRATE IONS: NO3-807 AND NO3-808, BUT NO ALTERNATE POSITIONS WERE MODELLED. ALTHOUGH THERE ARE SOME BUMPS BETWEEN ATOMS OF THE STRUCTURE, THESE ATOMS ARE CLEARLY DEFINED INTO THE ELECTRON DENSITIES.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.19 Å / Luzzati d res low obs: 5 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.45→1.52 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.853 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
