 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lzn | ||||||
|---|---|---|---|---|---|---|---|
| Title | NEUTRON STRUCTURE OF HEN EGG-WHITE LYSOZYME | ||||||
 Components Components | PROTEIN (LYSOZYME) | ||||||
 Keywords Keywords | HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method | NEUTRON DIFFRACTION / MOLECULAR REPLACEMEN XPLOR / Resolution: 1.7 Å | ||||||
 Authors Authors | Bon, C.I. / Lehmann, M.S. / Wilkinson, C. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 1999 Title: Quasi-Laue neutron-diffraction study of the water arrangement in crystals of triclinic hen egg-white lysozyme. Authors: Bon, C. / Lehmann, M.S. / Wilkinson, C. #1: Journal: Physica B (AMSTERDAM) / Year: 1998Title: Neutron Laue Diffraction in Macromolecular Crystallography Authors: Myles, D.A.A. / Bon, C. / Langan, P. / Cipriani, F. / Castagna, J.C. / Lehmann, M.S. / Wilkinson, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lzn.cif.gz | 72.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lzn.ent.gz | 53.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1lzn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lz/1lznftp://data.pdbj.org/pub/pdb/validation_reports/lz/1lzn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4lztS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Details: NITRATE IONS PRESENT / Source: (natural) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-NO3 /   Mass: 62.005 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: NO3 Mass: 62.005 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: NO3#3: Chemical | ChemComp-NA / |   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#4: Chemical | ChemComp-DOD / | 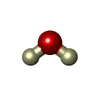  Mass: 18.015 Da / Num. of mol.: 243 / Source method: isolated from a natural source / Formula: D2O Mass: 18.015 Da / Num. of mol.: 243 / Source method: isolated from a natural source / Formula: D2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: NEUTRON DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.82 Å3/Da / Density % sol: 31.82 % Description: EXCHANGEABLE HYDROGENS (LABIL HYDROGENS+HYDROGENS WITHIN THE SOLVENT) WERE REPLACED BY DEUTERIUM. SITES OF EXCHANGEABLE HYDROGEN ON THE PROTEIN WERE ALL NAMED D*, AND WERE ASSIGNED THE ...Description: EXCHANGEABLE HYDROGENS (LABIL HYDROGENS+HYDROGENS WITHIN THE SOLVENT) WERE REPLACED BY DEUTERIUM. SITES OF EXCHANGEABLE HYDROGEN ON THE PROTEIN WERE ALL NAMED D*, AND WERE ASSIGNED THE SCATTERING LENGTH OF DEUTERIUM. WHEN THE HYDROGEN IS ACTUALLY EXCHANGED BY DEUTERIUM, THE OCCUPANCY IS 1, AND WHEN IT IS ACTUALLY NOT EXCHANGED,THE OCCUPANCY IS -0.56. PART OF SITES OF HYDRATION WERE MODELIZED AS COMPLETE D2O ENTITIES (MOLECULE DOD), AND PART OF THEM WERE MODELIZED AS SINGLE OXYGEN ATOM (MOLECULE HOH), BECAUSE OF DYNAMICAL DISORDER. | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.7 Details: BATCH TECHNIQUE EQUAL AMOUNT OF 1.0 % SOLUTION OF HEN EGG-WHITE LYSOZYME ( BOEHRINGER) AND 2.8 % NANO3 IN 50 MM ACETATE BUFFER (PH 4.7) | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: batch method | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 295 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Wavelength: 2.7-3.5 | |||||||||
| Detector | Detector: IMAGE PLATE / Date: Dec 1, 1997 | |||||||||
| Radiation | Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: neutron | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.7→13.6 Å / Num. obs: 8976 / % possible obs: 83 % / Observed criterion σ(I): 2 / Redundancy: 3.7 % / Rmerge(I) obs: 0.146 / Rsym value: 0.114 / Net I/σ(I): 35 | |||||||||
| Reflection | *PLUS Observed criterion σ(F): 2 / Num. measured all: 46061 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMEN XPLOR Starting model: PDB ENTRY 4LZT Resolution: 1.7→13.6 Å / Data cutoff high absF: 10000 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 Details: POSITIONS OF NON HYDROGEN ATOMS OF THE PROTEIN WERE KEPT FROM 4LZT, BECAUSE NEUTRON REFINEMENT CAN'T PROVIDE BETTER POSITIONS . WE BETTER FOCUSED OUR ATTENTION ON MODELING HYDROGEN AND HYDRATION SITES
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.82 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→13.6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: PARALLH22X.PRO / Topol file: TOPALLH22X | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name:  X-PLOR / Version: 3.1 / Classification: refinement X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.7 Å / Lowest resolution: 13.6 Å / σ(F): 2 / % reflection Rfree: 5 % / Rfactor obs: 0.204 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
