Green Fluorescent Protein / Green fluorescent protein / Green fluorescent protein, GFP / Green fluorescent protein-related / Green fluorescent protein / Green fluorescent protein / Beta Barrel / Mainly Beta Similarity search - Domain/homology
SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 11-STRANDED BARREL THIS IS REPRESENTED BY A 12-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
Mass: 18.015 Da / Num. of mol.: 318 / Source method: isolated from a natural source / Formula: H2O
Compound details
ENGINEERED RESIDUE IN CHAIN A, PHE 64 TO LEU ENGINEERED RESIDUE IN CHAIN A, GLN 80 TO ARG ...ENGINEERED RESIDUE IN CHAIN A, PHE 64 TO LEU ENGINEERED RESIDUE IN CHAIN A, GLN 80 TO ARG ENGINEERED RESIDUE IN CHAIN A, ILE 167 TO THR ENGINEERED RESIDUE IN CHAIN A, LYS 238 TO ASN
Has protein modification
Y
Nonpolymer details
GYS: THE CHROMOPHORE (GYS) IS FORMED FROM SER 65 - TYR 66 - GLY 67 BY CYCLIZATION OF THE ...GYS: THE CHROMOPHORE (GYS) IS FORMED FROM SER 65 - TYR 66 - GLY 67 BY CYCLIZATION OF THE POLYPEPTIDE BACKBONE BETWEEN NITROGEN OF GLY 67 AND CARBONYL CARBON OF SER 65. SUBSEQUENTLY THE CARBONYL OXYGEN IS ELIMINATED AS WATER AND TYR 66 IS OXIDIZED TO DEHYDROTYROSINE.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2 Å3/Da / Density % sol: 39 % / Description: SAD WAS BASED ON THE SULFUR SIGNAL UP TO 1.5 A
Crystal grow
Method: vapor diffusion, hanging drop / pH: 8 Details: HANGING DROP VAPOR DIFFUSION: 2 UL PROTEIN (10 MG/ML IN 20 MM TRIS, PH 8.0) PLUS 2 UL RESERVOIR (40% ETHANOL, 10 % DIOXANE)
Type: BRUKER SMART 6000 / Detector: CCD / Date: Aug 20, 2004 / Details: MONTEL MULTILAYER OPTIC
Radiation
Monochromator: MIRROR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.5418 Å / Relative weight: 1
Reflection
Resolution: 0.9→68 Å / Num. obs: 155426 / % possible obs: 91.4 % / Observed criterion σ(I): 0 / Redundancy: 19.7 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 20.35
Reflection shell
Resolution: 0.9→1 Å / Redundancy: 9.2 % / Rmerge(I) obs: 0.49 / Mean I/σ(I) obs: 3 / % possible all: 90.3
-
Processing
Software
Name
Version
Classification
SHELXL-97
refinement
SAINT
datareduction
SADABS
datascaling
SHELXD
SHELXE
phasing
Refinement
Method to determine structure: SAD Starting model: NONE Resolution: 0.9→6 Å / Num. parameters: 20655 / Num. restraintsaints: 26011 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 0.03.THERE ARE MANY WEAK REFLECTIONS WHICH ARE REMOVED IN THE CALCULATION OF REFLECTIONS WITH F>4SIG(F). THERE ARE MORE REFLECTIONS WITH ...Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 0.03.THERE ARE MANY WEAK REFLECTIONS WHICH ARE REMOVED IN THE CALCULATION OF REFLECTIONS WITH F>4SIG(F). THERE ARE MORE REFLECTIONS WITH F>0SIG(F) NUMBERING OF THE WATER MOLECULES IS SHIFTED BY 2000 COMPARED TO THE NUMBERS USED IN THE REFERENCE LISTED IN JRNL.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.174
4231
3 %
RANDOM
all
0.1456
133320
-
-
obs
-
-
90.3 %
-
Solvent computation
Solvent model: MOEWS & KRETSINGER
Refine analyze
Num. disordered residues: 29 / Occupancy sum hydrogen: 1814.6 / Occupancy sum non hydrogen: 2160.4
Refinement step
Cycle: LAST / Resolution: 0.9→6 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1821
0
25
318
2164
Refine LS restraints
Refine-ID
Type
Dev ideal
X-RAY DIFFRACTION
s_bond_d
0.034
X-RAY DIFFRACTION
s_angle_d
0.052
X-RAY DIFFRACTION
s_similar_dist
X-RAY DIFFRACTION
s_from_restr_planes
0.0329
X-RAY DIFFRACTION
s_zero_chiral_vol
0.145
X-RAY DIFFRACTION
s_non_zero_chiral_vol
0.148
X-RAY DIFFRACTION
s_anti_bump_dis_restr
X-RAY DIFFRACTION
s_rigid_bond_adp_cmpnt
0.015
X-RAY DIFFRACTION
s_similar_adp_cmpnt
0.042
X-RAY DIFFRACTION
s_approx_iso_adps
0.087
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 AEQUOREA VICTORIA (jellyfish)
AEQUOREA VICTORIA (jellyfish) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









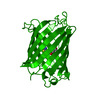


































































 PDBj
PDBj







 Assembly
Assembly


 Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O
Mass: 60.095 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O
 Mass: 46.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O
Mass: 46.068 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C2H6O Mass: 18.015 Da / Num. of mol.: 318 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 318 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing