 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2ykn: Crystal structure of HIV-1 Reverse Transcriptase (RT) in complex ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2ykn | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of HIV-1 Reverse Transcriptase (RT) in complex with a Difluoromethylbenzoxazole (DFMB) Pyrimidine Thioether derivative, a non-nucleoside RT inhibitor (NNRTI) | ||||||
 Components Components | (REVERSE TRANSCRIPTASE/RIBONUCLEASE H) x 2 | ||||||
 Keywords Keywords | HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |  HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 BH10 HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 BH10 | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.12 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.12 Å | ||||||
 Authors Authors | Boyer, J. / Arnoult, E. / Medebielle, M. / Guillemont, J. / Unge, T. / Unge, J. / Jochmans, D. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2011 Title: Difluoromethylbenzoxazole Pyrimidine Thioether Derivatives: A Novel Class of Potent Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. Authors: Boyer, J. / Arnoult, E. / Medebielle, M. / Guillemont, J. / Unge, J. / Jochmans, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2ykn.cif.gz | 214.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2ykn.ent.gz | 171.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2ykn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yk/2yknftp://data.pdbj.org/pub/pdb/validation_reports/yk/2ykn | HTTPS FTP |
|---|
-Related structure data

















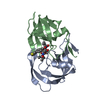































































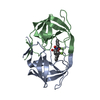

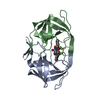






































-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 64790.246 Da / Num. of mol.: 1 / Fragment: RESIDUES 600-1156 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 BH10Production host:  References: UniProt: P03366, RNA-directed DNA polymerase, DNA-directed DNA polymerase, retroviral ribonuclease H |
|---|---|
| #2: Protein | Mass: 50039.488 Da / Num. of mol.: 1 / Fragment: RESIDUES 600-1027 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HUMAN IMMUNODEFICIENCY VIRUS TYPE 1 BH10Production host: References: UniProt: P03366, RNA-directed DNA polymerase, DNA-directed DNA polymerase, retroviral ribonuclease H |
| #3: Chemical | ChemComp-YKN / 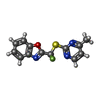  Mass: 293.292 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H9F2N3OS Mass: 293.292 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C13H9F2N3OS |
| #4: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 276 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 276 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | ENGINEERED |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.08 Å3/Da / Density % sol: 60.06 % / Description: NONE |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I911-2 / Wavelength: 1.0379 / Beamline: I911-2 / Wavelength: 1.0379 |
| Detector | Type: MARRESEARCH SX-165 / Detector: CCD / Date: Jun 11, 2008 / Details: MIRROR |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0379 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→19.45 Å / Num. obs: 74751 / % possible obs: 91 % / Observed criterion σ(I): 0 / Redundancy: 5.7 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 7.6 |
| Reflection shell | Resolution: 2.1→2.21 Å / Redundancy: 4.8 % / Rmerge(I) obs: 0.47 / Mean I/σ(I) obs: 1.7 / % possible all: 75 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: REVERSE TRANSCRIPTASE Resolution: 2.12→30 Å / Cor.coef. Fo:Fc: 0.939 / Cor.coef. Fo:Fc free: 0.912 / SU B: 5.44 / SU ML: 0.146 / Cross valid method: THROUGHOUT / ESU R: 0.233 / ESU R Free: 0.209 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.523 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.12→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|