 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ebk: Structural and kinetic analysis of drug resistant mutants of HIV-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ebk | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural and kinetic analysis of drug resistant mutants of HIV-1 protease | ||||||
 Components Components | HIV-1 PROTEASE | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / HIV-1 PROTEASE / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 2.06 Å X-RAY DIFFRACTION / Resolution: 2.06 Å | ||||||
 Authors Authors | Mahalingam, B. / Louis, J.M. / Reed, C.C. / Adomat, J.M. / Krouse, J. / Wang, Y.F. / Harrison, R.W. / Weber, I.T. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 1999 Title: Structural and kinetic analysis of drug resistant mutants of HIV-1 protease. Authors: Mahalingam, B. / Louis, J.M. / Reed, C.C. / Adomat, J.M. / Krouse, J. / Wang, Y.F. / Harrison, R.W. / Weber, I.T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ebk.cif.gz | 92.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ebk.ent.gz | 70.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1ebk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eb/1ebkftp://data.pdbj.org/pub/pdb/validation_reports/eb/1ebk | HTTPS FTP |
|---|
-Related structure data

















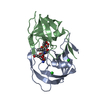



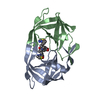

-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a dimer consisting of chains C and D or chains E and F |
-Components
| #1: Protein | Mass: 10711.611 Da / Num. of mol.: 4 / Fragment: FRAGMENT 69-167 / Mutation: Q7K, R8Q, L33I, L63I, C67A, C95A Source method: isolated from a genetically manipulated source Details: ENGINEERED MUTATIONS Q7K, R8Q, L33I, L63I, C67A, C95A STABILIZE THE PROTEASE FROM AUTOPROTEOLYSIS, WHILE RETAINING ACTIVITY SIMILAR TO WILD-TYPE HIV-1 PROTEASE Source: (gene. exp.) Human immunodeficiency virus 1 / Production host:  #2: Chemical | 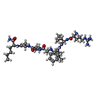  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 833.053 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C40H70N11O8 Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 833.053 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C40H70N11O8References: N-[(2R)-2-({N~5~-[amino(iminio)methyl]-L-ornithyl-L-valyl}amino)-4-methylpentyl]-L-phenylalanyl-L-alpha-glutamyl- L-alanyl-L-norleucinamide #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 144 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 144 / Source method: isolated from a natural source / Formula: H2ONonpolymer details | PEPTIDE INHIBITOR 0Q4 HAS A REDUCED PEPTIDE (-CH2-NH) INSTEAD OF THE NORMAL PEPTIDE LINK (-CO-NH). | Sequence details | ENGINEERED MUTATIONS Q7K, R8Q, L33I, L63I, C67A, C95A STABILIZE THE PROTEASE FROM AUTOPROTEOLYSIS, ...ENGINEERED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.58 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop Details: CITRATE/PHOSPHATE BUFFER 0.05M, DTT 10MM, DMSO 10%, SATURATED AMMONIUM SULPHATE (25-50%), PROTEIN 2-5 MG/ML, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion / PH range low: 5.2 / PH range high: 4.7 | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54 |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Num. obs: 19007 / % possible obs: 79.2 % / Rmerge(I) obs: 0.084 |
- Processing
Processing
| Software | Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.06→8 Å / Stereochemistry target values: Engh & Huber /
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.06→8 Å
| ||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 16.55 Å2 | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
