 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ebw | ||||||
|---|---|---|---|---|---|---|---|
| Title | HIV-1 protease in complex with the inhibitor BEA322 | ||||||
 Components Components | HIV-1 PROTEASE | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / Dimer / protein-inhibitor complex / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationHIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding ...HIV-1 retropepsin / symbiont-mediated activation of host apoptosis / retroviral ribonuclease H / exoribonuclease H / exoribonuclease H activity / DNA integration / viral genome integration into host DNA / establishment of integrated proviral latency / RNA-directed DNA polymerase / RNA stem-loop binding / viral penetration into host nucleus / host multivesicular body / RNA-directed DNA polymerase activity / RNA-DNA hybrid ribonuclease activity / Transferases; Transferring phosphorus-containing groups; Nucleotidyltransferases / host cell / viral nucleocapsid / DNA recombination / DNA-directed DNA polymerase / aspartic-type endopeptidase activity / Hydrolases; Acting on ester bonds / DNA-directed DNA polymerase activity / symbiont-mediated suppression of host gene expression / viral translational frameshifting / symbiont entry into host cell / lipid binding / host cell nucleus / host cell plasma membrane / virion membrane / structural molecule activity / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.81 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.81 Å | ||||||
 Authors Authors | Unge, T. | ||||||
 Citation Citation | Journal: Eur.J.Biochem. / Year: 2003 Title: Optimization of P1-P3 groups in symmetric and asymmetric HIV-1 protease inhibitors Authors: Andersson, H.O. / Fridborg, K. / Lowgren, S. / Alterman, M. / Muhlman, A. / Bjorsne, M. / Garg, N. / Kvarnstrom, I. / Schaal, W. / Classon, B. / Karlen, A. / Danielsson, U.H. / Ahlsen, G. / ...Authors: Andersson, H.O. / Fridborg, K. / Lowgren, S. / Alterman, M. / Muhlman, A. / Bjorsne, M. / Garg, N. / Kvarnstrom, I. / Schaal, W. / Classon, B. / Karlen, A. / Danielsson, U.H. / Ahlsen, G. / Nillroth, U. / Vrang, L. / Oberg, B. / Samuelsson, B. / Hallberg, A. / Unge, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ebw.cif.gz | 54.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ebw.ent.gz | 38.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1ebw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/eb/1ebwftp://data.pdbj.org/pub/pdb/validation_reports/eb/1ebw | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1d4hC  1d4iC  1d4jC  1ebyC  1ebzC  1ec1C  1ec2C  1ec3C C: citing same article ( |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a dimer |
-Components
| #1: Protein | Mass: 10803.756 Da / Num. of mol.: 2 / Fragment: FRAGMENT 69-167 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus 1 / Genus: Lentivirus / Plasmid: PET11C / Production host:  #2: Chemical | ChemComp-BEI / | 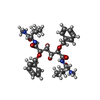  Mass: 642.783 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H50N4O8 Mass: 642.783 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H50N4O8#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 121 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.79 Å3/Da / Density % sol: 55.86 % | |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: 0.4 M Sodium chloride, 0.05 M MES, 0.02 % (W/V) sodium azide, pH 5.5, VAPOR DIFFUSION, HANGING DROP, temperature 277K | |||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 6.5 / Details: used microseeding | |||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.92 / Beamline: PX9.6 / Wavelength: 0.92 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 4, 1996 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.92 Å / Relative weight: 1 |
| Reflection | Resolution: 1.81→24.64 Å / Num. all: 22825 / Num. obs: 16594 / % possible obs: 72.8 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 2.7 % / Biso Wilson estimate: 10.5 Å2 / Rmerge(I) obs: 0.069 / Net I/σ(I): 12.7 |
| Reflection shell | Resolution: 1.81→1.92 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.186 / Num. unique all: 1324 / % possible all: 37.7 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Num. obs: 16613 / % possible obs: 69.7 % / Num. measured all: 42993 / Rmerge(I) obs: 0.094 |
| Reflection shell | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 1.86 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.81→24.64 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1329075.08 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber / Details: Refined with CNS program system
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 44.29 Å2 / ksol: 0.355 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.5 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.81→24.64 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.81→1.92 Å / Rfactor Rfree error: 0.028 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 24.6 Å / Rfactor Rfree: 0.218 / Rfactor Rwork: 0.19 | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.23 |
