 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3mb4: Crystal Structure of the fifth Bromodomain of Human Poly-bromodom... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3mb4 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the fifth Bromodomain of Human Poly-bromodomain containing protein 1 (PB1) with NMP | ||||||
 Components Components | Protein polybromo-1 | ||||||
 Keywords Keywords | TRANSCRIPTION / PB1 / polybromo 1 isoform 1 / BAF180 / Polybromo-1D / PBRM1 / BRG1-associated factor 180 / Structural Genomics Consortium / SGC / Bromodomain / Chromatin regulator / DNA-binding / Nucleus / Phosphoprotein / Transcription regulation | ||||||
| Function / homology |  Function and homology information Function and homology informationFormation of the polybromo-BAF (pBAF) complex / regulation of G0 to G1 transition / RSC-type complex / regulation of nucleotide-excision repair / regulation of mitotic metaphase/anaphase transition / SWI/SNF complex / positive regulation of T cell differentiation / nuclear chromosome / positive regulation of double-strand break repair / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known ...Formation of the polybromo-BAF (pBAF) complex / regulation of G0 to G1 transition / RSC-type complex / regulation of nucleotide-excision repair / regulation of mitotic metaphase/anaphase transition / SWI/SNF complex / positive regulation of T cell differentiation / nuclear chromosome / positive regulation of double-strand break repair / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / positive regulation of myoblast differentiation / regulation of G1/S transition of mitotic cell cycle / positive regulation of cell differentiation / transcription elongation by RNA polymerase II / kinetochore / nuclear matrix / RMTs methylate histone arginines / mitotic cell cycle / chromatin remodeling / negative regulation of cell population proliferation / chromatin binding / regulation of transcription by RNA polymerase II / chromatin / DNA binding / nucleoplasm / nucleus Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.66 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.66 Å | ||||||
 Authors Authors | Filippakopoulos, P. / Picaud, S. / Keates, T. / Ugochukwu, E. / von Delft, F. / Arrowsmith, C.H. / Edwards, A.M. / Weigelt, J. / Bountra, C. / Knapp, S. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2012 Title: Histone recognition and large-scale structural analysis of the human bromodomain family. Authors: Filippakopoulos, P. / Picaud, S. / Mangos, M. / Keates, T. / Lambert, J.P. / Barsyte-Lovejoy, D. / Felletar, I. / Volkmer, R. / Muller, S. / Pawson, T. / Gingras, A.C. / Arrowsmith, C.H. / Knapp, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3mb4.cif.gz | 117 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3mb4.ent.gz | 91.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3mb4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mb/3mb4ftp://data.pdbj.org/pub/pdb/validation_reports/mb/3mb4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2nxbC  2oo1C  2ossC  2ouoC  2rfjC  3d7cC  3daiC  3dwyC  3gg3C  3hmeC  3hmfC  3hmhC  3i3jC  3iu5C  3iu6C  3lxjC  3mb3C  3mqmC  3nxbC  3p1cC  3p1dC  3q2eC  3rcwC  3tlpC  3uv2C  3uv4C  3uv5C  3uvdC  3uvwC  3uvxC  3uvyC  3uw9C  3g0jS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14648.000 Da / Num. of mol.: 2 / Fragment: UNP residues 645-766 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: BAF180, PB1, PBRM1 / Plasmid: pNIC28-Bsa4 / Production host:  #2: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C2H6O2#3: Chemical | 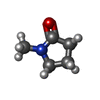  Mass: 99.131 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H9NO Mass: 99.131 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C5H9NO#4: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cl#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51.26 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 0.15M NaNO3 25% PEG 3350 10% Ethylene Glycol, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E SUPERBRIGHT / Wavelength: 1.5 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Nov 2, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5 Å / Relative weight: 1 |
| Reflection | Resolution: 1.66→50 Å / Num. all: 36073 / Num. obs: 36037 / % possible obs: 99.9 % / Redundancy: 7.2 % / Rmerge(I) obs: 0.067 / Rsym value: 0.064 / Net I/σ(I): 25 |
| Reflection shell | Resolution: 1.66→1.72 Å / Redundancy: 6.6 % / Rmerge(I) obs: 0.955 / Mean I/σ(I) obs: 1.97 / Num. unique all: 3536 / Rsym value: 0.817 / % possible all: 99.8 |
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Rfactor: 46.4 / Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 3G0J Resolution: 1.66→19.01 Å / Cor.coef. Fo:Fc: 0.966 / Cor.coef. Fo:Fc free: 0.959 / WRfactor Rfree: 0.243 / WRfactor Rwork: 0.215 / Occupancy max: 1 / Occupancy min: 0.3 / FOM work R set: 0.853 / SU B: 4.137 / SU ML: 0.072 / SU R Cruickshank DPI: 0.097 / SU Rfree: 0.093 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.097 / ESU R Free: 0.093 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES: WITH TLS ADDED
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 103.33 Å2 / Biso mean: 37.169 Å2 / Biso min: 19.32 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.66→19.01 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.66→1.703 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
