Entry Database : PDB / ID : 2vdbTitle Structure of human serum albumin with S-naproxen and the GA module PEPTOSTREPTOCOCCAL ALBUMIN-BINDING PROTEIN SERUM ALBUMIN Keywords / / / / / / / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species PEPTOSTREPTOCOCCUS MAGNUS (bacteria)HOMO SAPIENS (human)Method / / / Resolution : 2.52 Å Authors Lejon, S. / Cramer, J.F. / Nordberg, P.A. Journal : Acta Crystallogr.,Sect.F / Year : 2008Title : Structural Basis for the Binding of Naproxen to Human Serum Albumin in the Presence of Fatty Acids and the Ga Module.Authors : Lejon, S. / Cramer, J.F. / Nordberg, P.A. History Deposition Oct 4, 2007 Deposition site / Processing site Revision 1.0 Feb 26, 2008 Provider / Type Revision 1.1 Jul 13, 2011 Group / Version format complianceRevision 1.2 Dec 13, 2023 Group Data collection / Database references ... Data collection / Database references / Other / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sfRevision 1.3 Oct 23, 2024 Group / Category / pdbx_modification_feature / Item
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information PEPTOSTREPTOCOCCUS MAGNUS (bacteria)
PEPTOSTREPTOCOCCUS MAGNUS (bacteria) HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

























































 PDBj
PDBj











 Assembly
Assembly
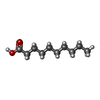
 Mass: 172.265 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H20O2
Mass: 172.265 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H20O2
 Mass: 230.259 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H14O3 / Comment: antiinflammatory*YM
Mass: 230.259 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H14O3 / Comment: antiinflammatory*YM Sample preparation
Sample preparation / Beamline: ID14-3 / Wavelength: 0.931
/ Beamline: ID14-3 / Wavelength: 0.931  Processing
Processing