 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ha2: Human Serum Albumin Complexed With Myristic Acid and the S-(-) en... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ha2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human Serum Albumin Complexed With Myristic Acid and the S-(-) enantiomer of warfarin | ||||||
 Components Components | SERUM ALBUMIN | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / SERUM PROTEIN / DRUG BINDING / ANTI-COAGULANT | ||||||
| Function / homology |  Function and homology information Function and homology informationbilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / Prednisone ADME / Heme degradation ...bilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / Prednisone ADME / Heme degradation / molecular carrier activity / Aspirin ADME / antioxidant activity / Scavenging of heme from plasma / toxic substance binding / Recycling of bile acids and salts / platelet alpha granule lumen / fatty acid binding / cellular response to starvation / Post-translational protein phosphorylation / Cytoprotection by HMOX1 / response to nutrient levels / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / Platelet degranulation / pyridoxal phosphate binding / protein-folding chaperone binding / blood microparticle / endoplasmic reticulum lumen / copper ion binding / Golgi apparatus / endoplasmic reticulum / protein-containing complex / DNA binding / : / extracellular exosome / extracellular region / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Petitpas, I. / Bhattacharya, A.A. / Curry, S. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: Crystal Structure Analysis of Warfarin Binding to Human Serum Albumin: Anatomy of Drug Site I Authors: Petitpas, I. / Bhattacharya, A.A. / Twine, S. / East, M. / Curry, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ha2.cif.gz | 128.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ha2.ent.gz | 100.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1ha2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ha/1ha2ftp://data.pdbj.org/pub/pdb/validation_reports/ha/1ha2 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1h9zC  1e7gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

































-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 66571.219 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-MYR / 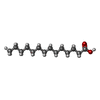  Mass: 228.371 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C14H28O2 Mass: 228.371 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C14H28O2#3: Chemical | ChemComp-SWF / | 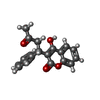  Mass: 308.328 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H16O4 Mass: 308.328 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H16O4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 23 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Sequence details | PROTEIN HAS A 24 AA LEADER NOT PRESENT IN THE CRYSTAL | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 60 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 / Details: SEE PUBLICATION, pH 7.00 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop / Details: Curry, S., (1998) Nat. Struct. Biol., 5, 827. / pH: 7.5 | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.91 / Beamline: X11 / Wavelength: 0.91 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jul 15, 2000 / Details: MIRRORS |
| Radiation | Monochromator: TRIANGULAR MONOCHROMATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.91 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→30 Å / Num. obs: 23090 / % possible obs: 98.4 % / Redundancy: 1.9 % / Biso Wilson estimate: 60 Å2 / Rmerge(I) obs: 0.065 / Net I/σ(I): 4.4 |
| Reflection shell | Resolution: 2.5→2.64 Å / Redundancy: 2 % / Rmerge(I) obs: 0.314 / Mean I/σ(I) obs: 1.8 / % possible all: 98.4 |
| Reflection shell | *PLUS % possible obs: 98.2 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1E7G Resolution: 2.5→29.45 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 1559741.98 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Stereochemistry target values: MAXIMUM LIKELIHOOD USING AMPLITUDES
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 39.5591 Å2 / ksol: 0.282755 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 63.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→29.45 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.66 Å / Rfactor Rfree error: 0.029 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
