 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1hk4: HUMAN SERUM ALBUMIN COMPLEXED WITH THYROXINE (3,3',5,5'-TETRAIODO... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1hk4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | HUMAN SERUM ALBUMIN COMPLEXED WITH THYROXINE (3,3',5,5'-TETRAIODO-L-THYRONINE) and myristic acid (tetradecanoic acid) | |||||||||
 Components Components | SERUM ALBUMIN | |||||||||
 Keywords Keywords | PLASMA PROTEIN / HORMONE-BINDING / LIPID-BINDING / THYROXINE / FAMILIAL DYSALBUMINEMIC HYPERTHYROXINEMIA | |||||||||
| Function / homology |  Function and homology information Function and homology informationbilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / molecular carrier activity / Prednisone ADME ...bilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / molecular carrier activity / Prednisone ADME / Heme degradation / Aspirin ADME / antioxidant activity / Scavenging of heme from plasma / toxic substance binding / Recycling of bile acids and salts / platelet alpha granule lumen / fatty acid binding / cellular response to starvation / Post-translational protein phosphorylation / Cytoprotection by HMOX1 / response to nutrient levels / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / Platelet degranulation / pyridoxal phosphate binding / protein-folding chaperone binding / blood microparticle / endoplasmic reticulum lumen / copper ion binding / Golgi apparatus / endoplasmic reticulum / protein-containing complex / : / DNA binding / extracellular exosome / extracellular region / identical protein binding / nucleus Similarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | |||||||||
 Authors Authors | Petitpas, I. / Petersen, C.E. / Ha, C.E. / Bhattacharya, A.A. / Zunszain, P.A. / Ghuman, J. / Bhagavan, N.V. / Curry, S. | |||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2003 Title: Structural Basis of Albumin-Thyroxine Interactions and Familial Dysalbuminemic Hyperthyroxinemia Authors: Petitpas, I. / Petersen, C.E. / Ha, C.E. / Bhattacharya, A.A. / Zunszain, P.A. / Ghuman, J. / Bhagavan, N.V. / Curry, S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1hk4.cif.gz | 128.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1hk4.ent.gz | 98.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1hk4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hk/1hk4ftp://data.pdbj.org/pub/pdb/validation_reports/hk/1hk4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1hk1C  1hk2C  1hk3C  1hk5C  1e7gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




























-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 66571.219 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-MYR / 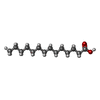  Mass: 228.371 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C14H28O2 Mass: 228.371 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C14H28O2#3: Chemical | ChemComp-T44 / |   Mass: 776.870 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H11I4NO4 / Comment: hormone*YM Mass: 776.870 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C15H11I4NO4 / Comment: hormone*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 15 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 15 / Source method: isolated from a natural source / Formula: H2OCompound details | SERUM ALBUMIN, REGULATES THE COLLOIDAL OSMOTIC PRESSURE OF WATER, IT BINDS TO CA++, NA+, K+, FATTY ...SERUM ALBUMIN, REGULATES THE COLLOIDAL OSMOTIC PRESSURE OF WATER, IT BINDS TO CA++, NA+, K+, FATTY ACIDS, HORMONES, BILIRUBIN AND DRUGS | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 54 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 / Details: pH 7.00 | ||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop / PH range low: 7.5 / PH range high: 7 | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jan 15, 1999 / Details: MIRRORS |
| Radiation | Monochromator: SI CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→37.5 Å / Num. obs: 24201 / % possible obs: 90.6 % / Observed criterion σ(I): 0 / Redundancy: 2.1 % / Biso Wilson estimate: 39.9 Å2 / Rmerge(I) obs: 0.079 / Net I/σ(I): 5.6 |
| Reflection shell | Resolution: 2.4→2.53 Å / Redundancy: 1.7 % / Rmerge(I) obs: 0.221 / Mean I/σ(I) obs: 2.7 / % possible all: 76 |
| Reflection shell | *PLUS % possible obs: 76 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1E7G Resolution: 2.4→37.24 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 1776051.07 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: BULK SOLVENT MODEL USED RESIDUES 1-2 AND 585 ARE DISORDERED AND HAVE NOT BEEN INCLUDED IN THE MODEL.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 50.9549 Å2 / ksol: 0.323902 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 52.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→37.24 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.027 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 37.5 Å / % reflection Rfree: 5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
