 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2bxk: Human serum albumin complexed with myristate, azapropazone and in... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bxk | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human serum albumin complexed with myristate, azapropazone and indomethacin | |||||||||
 Components Components | SERUM ALBUMIN | |||||||||
 Keywords Keywords | TRANSPORT PROTEIN / ALBUMIN / CARRIER PROTEIN / LIPID-BINDING / METAL-BINDING / DRUG-BINDING / AZAPROPAZONE / MYRISTATE / INDOMETHACIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationbilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / Prednisone ADME / Heme degradation ...bilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / Prednisone ADME / Heme degradation / molecular carrier activity / Aspirin ADME / antioxidant activity / Scavenging of heme from plasma / toxic substance binding / Recycling of bile acids and salts / platelet alpha granule lumen / fatty acid binding / cellular response to starvation / Post-translational protein phosphorylation / Cytoprotection by HMOX1 / response to nutrient levels / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / Platelet degranulation / pyridoxal phosphate binding / protein-folding chaperone binding / blood microparticle / endoplasmic reticulum lumen / copper ion binding / Golgi apparatus / endoplasmic reticulum / protein-containing complex / DNA binding / : / extracellular exosome / extracellular region / identical protein binding / nucleus Similarity search - Function | |||||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | |||||||||
 Authors Authors | Ghuman, J. / Zunszain, P.A. / Petitpas, I. / Bhattacharya, A.A. / Curry, S. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2005 Title: Structural Basis of the Drug-Binding Specificity of Human Serum Albumin. Authors: Ghuman, J. / Zunszain, P.A. / Petitpas, I. / Bhattacharya, A.A. / Otagiri, M. / Curry, S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bxk.cif.gz | 128.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bxk.ent.gz | 99.7 KB | Display | PDB format |
| PDBx/mmJSON format | 2bxk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bx/2bxkftp://data.pdbj.org/pub/pdb/validation_reports/bx/2bxk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2bx8C  2bxaC  2bxbC  2bxcC  2bxdC  2bxeC  2bxfC  2bxgC  2bxhC  2bxiC  2bxlC  2bxmC  2bxnC  2bxoC  2bxpC  2bxqC  1e7gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













































-Links
 PDBj
PDBj















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 66543.141 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Tissue: PLASMA / Production host:  PICHIA PASTORIS (fungus) / References: UniProt: P02768 PICHIA PASTORIS (fungus) / References: UniProt: P02768 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-MYR / 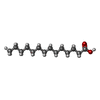  Mass: 228.371 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C14H28O2 Mass: 228.371 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C14H28O2#3: Chemical | ChemComp-IMN / | 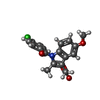  Mass: 357.788 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H16ClNO4 / Comment: medication, antiinflammatory*YM Mass: 357.788 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H16ClNO4 / Comment: medication, antiinflammatory*YM#4: Chemical | ChemComp-AZQ / | 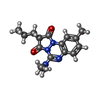  Mass: 300.356 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H20N4O2 Mass: 300.356 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H20N4O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Sequence details | SEQUENCE CONTAINS SIGNAL AND PRO-PEPTIDE (RESIDUES 1-24 IN DATABASE ENTRY). PROTEIN CRYSTALLISED ...SEQUENCE CONTAINS SIGNAL AND PRO-PEPTIDE (RESIDUES 1-24 IN DATABASE ENTRY). PROTEIN CRYSTALLIS | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.53 Å3/Da / Density % sol: 44 % |
|---|---|
| Crystal grow | pH: 7 / Details: pH 7.00 |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Oct 25, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→22.3 Å / Num. obs: 25123 / % possible obs: 95.1 % / Observed criterion σ(I): 0 / Redundancy: 2.6 % / Biso Wilson estimate: 48.2 Å2 / Rmerge(I) obs: 0.04 / Net I/σ(I): 13.6 |
| Reflection shell | Resolution: 2.4→2.53 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.32 / Mean I/σ(I) obs: 3.6 / % possible all: 95.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1E7G Resolution: 2.4→21.56 Å / Rfactor Rfree error: 0.008 / Data cutoff high absF: 1792393.36 / Isotropic thermal model: GROUP / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: MLF
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 55.3534 Å2 / ksol: 0.335879 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 50.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→21.56 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.036 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
