| Entry | Database: PDB / ID: 2clk
|
|---|
| Title | Tryptophan Synthase in complex with D-glyceraldehyde 3-phosphate (G3P) |
|---|
 Components Components | (TRYPTOPHAN SYNTHASE ...) x 2 |
|---|
 Keywords Keywords | LYASE / AROMATIC AMINO ACID BIOSYNTHESIS / TRYPTOPHAN BIOSYNTHESIS / CRBON- OXYGEN LYASE / AMINO-ACID BIOSYNTHESIS / ALLOSTERIC ENZYME / PYRIDOXAL PHOSPHATE |
|---|
| Function / homology |  Function and homology information Function and homology information
tryptophan synthase / tryptophan synthase activity / L-tryptophan biosynthetic process / identical protein binding / cytoplasm / cytosolSimilarity search - Function Tryptophan synthase, alpha chain / Tryptophan synthase, alpha chain, active site / Tryptophan synthase alpha chain / Tryptophan synthase alpha chain signature. / Tryptophan synthase, beta chain, conserved site / Tryptophan synthase, beta chain / Tryptophan synthase beta chain/beta chain-like / Tryptophan synthase beta chain pyridoxal-phosphate attachment site. / Rossmann fold - #1100 / Pyridoxal-phosphate dependent enzyme ...Tryptophan synthase, alpha chain / Tryptophan synthase, alpha chain, active site / Tryptophan synthase alpha chain / Tryptophan synthase alpha chain signature. / Tryptophan synthase, beta chain, conserved site / Tryptophan synthase, beta chain / Tryptophan synthase beta chain/beta chain-like / Tryptophan synthase beta chain pyridoxal-phosphate attachment site. / Rossmann fold - #1100 / Pyridoxal-phosphate dependent enzyme / Pyridoxal-phosphate dependent enzyme / Tryptophan synthase beta subunit-like PLP-dependent enzyme / Ribulose-phosphate binding barrel / Aldolase class I / Aldolase-type TIM barrel / TIM Barrel / Alpha-Beta Barrel / Rossmann fold / 3-Layer(aba) Sandwich / Alpha BetaSimilarity search - Domain/homology GLYCERALDEHYDE-3-PHOSPHATE / PYRIDOXAL-5'-PHOSPHATE / Tryptophan synthase alpha chain / Tryptophan synthase beta chainSimilarity search - Component |
|---|
| Biological species |  SALMONELLA TYPHIMURIUM (bacteria) SALMONELLA TYPHIMURIUM (bacteria) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å |
|---|
 Authors Authors | Ngo, H. / Harris, R. / Kimmich, N. / Casino, P. / Niks, D. / Blumenstein, L. / Barends, T.R. / Kulik, V. / Weyand, M. / Schlichting, I. / Dunn, M.F. |
|---|
 Citation Citation | Journal: Biochemistry / Year: 2007
Title: Synthesis and Characterization of Allosteric Probes of Substrate Channeling in the Tryptophan Synthase Bienzyme Complex.
Authors: Ngo, H. / Harris, R. / Kimmich, N. / Casino, P. / Niks, D. / Blumenstein, L. / Barends, T.R. / Kulik, V. / Weyand, M. / Schlichting, I. / Dunn, M.F. |
|---|
| History | | Deposition | Apr 27, 2006 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | Jun 12, 2007 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | May 8, 2011 | Group: Version format compliance |
|---|
| Revision 1.2 | Jul 13, 2011 | Group: Version format compliance |
|---|
| Revision 1.3 | Apr 9, 2025 | Group: Data collection / Database references ...Data collection / Database references / Derived calculations / Other / Structure summary
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_status / pdbx_entry_details / pdbx_struct_conn_angle / struct_conn
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_status.status_code_sf / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

































































 PDBj
PDBj

 Assembly
Assembly

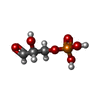



 Mass: 170.058 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7O6P
Mass: 170.058 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7O6P Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P
Mass: 247.142 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H10NO6P Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Sample preparation
Sample preparation / Beamline: ID14-1 / Wavelength: 0.934
/ Beamline: ID14-1 / Wavelength: 0.934  Processing
Processing