 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1wxj | ||||||
|---|---|---|---|---|---|---|---|
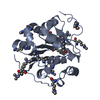
| タイトル | Crystal Structure Of Tryptophan Synthase A-Subunit with Indole-3-propanol phosphate From Thermus Thermophilus Hb8 | ||||||
 要素 要素 | tryptophan synthase alpha chain | ||||||
 キーワード キーワード | LYASE / structural genomics / Thermus thermophilus HB8 / RIKEN Structural Genomics/Proteomics Initiative / RSGI | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | ||||||
| 生物種 |   Thermus thermophilus (バクテリア) Thermus thermophilus (バクテリア) | ||||||
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.7 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.7 Å | ||||||
 データ登録者 データ登録者 | Asada, Y. / Kunishima, N. / RIKEN Structural Genomics/Proteomics Initiative (RSGI) | ||||||
 引用 引用 | ジャーナル: To be Published タイトル: Crystal Structure Of Tryptophan Synthase A-Subunit with Indole-3-propanol phosphate From Thermus Thermophilus Hb8 著者: Asada, Y. / Kunishima, N. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1wxj.cif.gz | 66.9 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1wxj.ent.gz | 47.2 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1wxj.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/wx/1wxjftp://data.pdbj.org/pub/pdb/validation_reports/wx/1wxj | HTTPS FTP |
|---|
-関連構造データ
| 関連構造データ |  1ujpS S: 精密化の開始モデル |
|---|---|
| 類似構造データ | |
| その他のデータベース |









-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
| ||||||||
| 詳細 | The biological assembly is monomer |
-要素
| #1: タンパク質 | 分子量: 28977.525 Da / 分子数: 1 / 由来タイプ: 組換発現 由来: (組換発現) Thermus thermophilus (バクテリア)株: HB8 / プラスミド: pET11a / 生物種 (発現宿主): Escherichia coli / 発現宿主: | ||||
|---|---|---|---|---|---|
| #2: 化合物 | ChemComp-SO4 /   分子量: 96.063 Da / 分子数: 4 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 4 / 由来タイプ: 合成 / 式: SO4#3: 化合物 | ChemComp-IPL / | 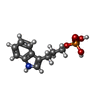  分子量: 255.207 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C11H14NO4P 分子量: 255.207 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C11H14NO4P#4: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 278 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 278 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.16 Å3/Da / 溶媒含有率: 43 % |
|---|---|
| 結晶化 | 温度: 291 K / 手法: マイクロバッチ法 / pH: 8.5 詳細: INDOLE-3-PROPANOL PHOSPHATE, Tris-HCl, Ammonium Sulfate, pH 8.5, MICROBATCH, temperature 291K |
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: SPring-8  / ビームライン: BL26B1 / 波長: 0.9 Å / ビームライン: BL26B1 / 波長: 0.9 Å |
| 検出器 | タイプ: RIGAKU JUPITER 210 / 検出器: CCD / 日付: 2004年4月23日 / 詳細: mirrors |
| 放射 | モノクロメーター: Bending Magnet / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 0.9 Å / 相対比: 1 |
| 反射 | 解像度: 1.7→50 Å / Num. all: 26921 / Num. obs: 26921 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / 冗長度: 3.8 % / Biso Wilson estimate: 18.927 Å2 / Rmerge(I) obs: 0.044 / Rsym value: 0.038 / Net I/σ(I): 15.1 |
| 反射 シェル | 解像度: 1.7→1.76 Å / 冗長度: 3.8 % / Rmerge(I) obs: 0.297 / Mean I/σ(I) obs: 4.01 / Num. unique all: 2664 / Rsym value: 0.253 / % possible all: 100 |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: 1UJP 解像度: 1.7→27.08 Å / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber
| |||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 23.3 Å2
| |||||||||||||||||||||||||
| Refine analyze |
| |||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.7→27.08 Å
| |||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.7→1.78 Å / Rfactor Rfree error: 0.022
|
