 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gqr | ||||||
|---|---|---|---|---|---|---|---|
| Title | ACETYLCHOLINESTERASE (E.C. 3.1.1.7) COMPLEXED WITH RIVASTIGMINE | ||||||
 Components Components | ACETYLCHOLINESTERASE | ||||||
 Keywords Keywords | HYDROLASE / NEUROTRANSMITTER CLEAVAGE | ||||||
| Function / homology |  Function and homology information Function and homology informationacetylcholine catabolic process in synaptic cleft / acetylcholinesterase / choline metabolic process / acetylcholinesterase activity / side of membrane / synaptic cleft / synapse / : / plasma membrane Similarity search - Function | ||||||
| Biological species |   TORPEDO CALIFORNICA (Pacific electric ray) TORPEDO CALIFORNICA (Pacific electric ray) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Raves, M.L. / Harel, M. / Silman, I. / Sussman, J.L. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2002 Title: Kinetic and Structural Studies on the Interaction of Cholinesterases with the Anti-Alzheimer Drug Rivastigmine Authors: Bar-on, P. / Millard, C.B. / Harel, M. / Dvir, H. / Enz, A. / Sussman, J.L. / Silman, I. #1: Journal: Biochem.Biophys.Res.Commun. / Year: 1992 Title: Mechanism of Inhibition of Cholinesterases by Huperzine A Authors: Ashani, Y. / Peggins III, J.O. / Doctor, B.P. #2: Journal: Acta Crystallogr.,Sect.C / Year: 1991 Title: Huperzine A--A Potent Acetylcholinesterase Inhibitor of Use in the Treatment of Alzheimer'S Disease Authors: Geib, S.J. / Tuckmantel, W. / Kozikowski, A.P. #3: Journal: Science / Year: 1991 Title: Atomic Structure of Acetylcholinesterase from Torpedo Californica: A Prototypic Acetylcholine-Binding Protein Authors: Sussman, J.L. / Harel, M. / Frolow, F. / Oefner, C. / Goldman, A. / Toker, L. / Silman, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gqr.cif.gz | 129 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gqr.ent.gz | 98.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1gqr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gq/1gqrftp://data.pdbj.org/pub/pdb/validation_reports/gq/1gqr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1gqsC  1ace C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |













































-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 60193.977 Da / Num. of mol.: 1 / Fragment: RESIDUES 25-556 / Source method: isolated from a natural source Source: (natural) TORPEDO CALIFORNICA (Pacific electric ray)Organ: ELECTRIC ORGAN / Variant: G2 FORM / Tissue: ELECTROPLAQUE / References: UniProt: P04058, acetylcholinesterase | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Sugar |   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #3: Chemical | ChemComp-SAF / | 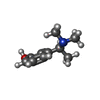  Mass: 165.232 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15NO Mass: 165.232 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15NO#4: Chemical | ChemComp-EMM / [ | 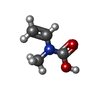  Mass: 101.104 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H7NO2 Mass: 101.104 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H7NO2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 386 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 386 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.12 Å3/Da / Density % sol: 70.13 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.8 / Details: pH 5.80 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 1.1 / Beamline: X12C / Wavelength: 1.1 |
| Detector | Date: Jul 15, 1997 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→30 Å / Num. obs: 50861 / % possible obs: 99.7 % / Observed criterion σ(I): 0 / Redundancy: 4.8 % / Biso Wilson estimate: 21.1 Å2 / Rmerge(I) obs: 0.066 / Net I/σ(I): 17.7 |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 2.6 % / Rmerge(I) obs: 0.322 / Mean I/σ(I) obs: 1.4 / % possible all: 97.1 |
| Reflection | *PLUS Num. all: 50861 / Num. obs: 46214 / Num. measured all: 242981 |
| Reflection shell | *PLUS Highest resolution: 2.2 Å / % possible obs: 97.1 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1ACE 1ace Resolution: 2.2→29.13 Å / Rfactor Rfree error: 0.005 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→29.13 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.34 Å / Rfactor Rfree error: 0.026 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: PARHCSDX.PRO / Topol file: TOPHCSDX.PRO | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 29.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
