 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1eve: THREE DIMENSIONAL STRUCTURE OF THE ANTI-ALZHEIMER DRUG, E2020 (AR... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eve | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | THREE DIMENSIONAL STRUCTURE OF THE ANTI-ALZHEIMER DRUG, E2020 (ARICEPT), COMPLEXED WITH ITS TARGET ACETYLCHOLINESTERASE | |||||||||
 Components Components | ACETYLCHOLINESTERASE | |||||||||
 Keywords Keywords | SERINE HYDROLASE / ALZHEIMER'S DISEASE / DRUG / ALPHA/BETA HYDROLASE / NEUROTRANSMITTER CLEAVAGE / CATALYTIC TRIAD / GLYCOSYLATED PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationacetylcholine catabolic process in synaptic cleft / acetylcholinesterase / choline metabolic process / acetylcholinesterase activity / side of membrane / synaptic cleft / synapse / : / plasma membrane Similarity search - Function | |||||||||
| Biological species |   Torpedo californica (Pacific electric ray) Torpedo californica (Pacific electric ray) | |||||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | |||||||||
 Authors Authors | Kryger, G. / Silman, I. / Sussman, J.L. | |||||||||
 Citation Citation | Journal: Structure Fold.Des. / Year: 1999 Title: Structure of acetylcholinesterase complexed with E2020 (Aricept): implications for the design of new anti-Alzheimer drugs. Authors: Kryger, G. / Silman, I. / Sussman, J.L. #1: Journal: Bioorg.Med.Chem. / Year: 1996Title: The Rationale for E2020 as a Potent Acetylcholinesterase Inhibitor Authors: Kawakami, Y. / Inoue, A. / Kawai, T. / Wakita, M. / Sugimoto, H. / Hopfinger, A.J. #2: Journal: Science / Year: 1991Title: Atomic Structure of Acetylcholinesterase from Torpedo Californica: A Prototypic Acetylcholine-Binding Protein Authors: Sussman, J.L. / Harel, M. / Frolow, F. / Oefner, C. / Goldman, A. / Toker, L. / Silman, I. | |||||||||
| History |
| |||||||||
| Remark 650 | HELIX THE ENZYME IS A GPI-ANCHORED DIMER, THE TWO MONOMERS IN THE DIMER ARE RELATED BY ...HELIX THE ENZYME IS A GPI-ANCHORED DIMER, THE TWO MONOMERS IN THE DIMER ARE RELATED BY CRYSTALLOGRAPHIC TWO-FOLD SYMMETRY AND GENERATE A FOUR HELIX BUNDLE A365-A375 AND A518-A535. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eve.cif.gz | 131.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eve.ent.gz | 101.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1eve.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ev/1eveftp://data.pdbj.org/pub/pdb/validation_reports/ev/1eve | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2aceS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | TORPEDO CALIFORNICA ACETYLCHOLINESTERASE IS A G2 DIMER IN SOLUTION (SEE SUSSMAN 1988). THE ASYMMETRIC UNIT CONTAINS A MONOMER, WITH THE CRYSTALLOGRAPHIC TWO-FOLD AXIS RELATING THE TWO MONOMERS IN A DIMER. THIS STRUCTURE IS MORE COMPLETE THAN THE STARTING MODEL OF THE NATIVE STRUCTURE (PDB ID 2ACE). RESIDUES THAT ARE NOT SEEN IN THE CRYSTAL STRUCTURE DUE TO DISORDER INCLUDE THE N-TERMINAL RESIDUE ASP 1 AND THE C-TERMINAL RESIDUES AFTER THR 535. THR 535 IS THE LAST RESIDUE OBSERVED AT THE C-TERMINUS. THE LIGAND SEEN IN THE STRUCTURE, E2020 (DONEPEZIL, ARICEPT), IS A POTENT REVERSIBLE ACHE INHIBITOR WHICH IS AN FDA APPROVED DRUG FOR THE SYMPTOMATIC TREATMENT OF ALZHEIMER'S DISEASE (SEE KAWAKAMI 1996). THE CHIRAL INHIBITOR WAS SOAKED AS A RACEMATE BUT ONLY THE R FORM SEEMS TO BIND TO THE ENZYME ACCORDING TO THE X-RAY DIFFRACTION EXPERIMENT. |
-Components
| #1: Protein | Mass: 61325.090 Da / Num. of mol.: 1 / Source method: isolated from a natural source Source: (natural) Torpedo californica (Pacific electric ray)Organ: ELECTRIC ORGAN / Variant: G2 FORM / Tissue: ELECTROPLAQUE / References: UniProt: P04058, acetylcholinesterase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source | ||||||
| #3: Sugar |   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 3 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 3Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #4: Chemical | ChemComp-E20 / | 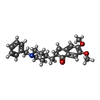  Mass: 379.492 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H29NO3 Mass: 379.492 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H29NO3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 396 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 396 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.8 Å3/Da / Density % sol: 68 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.8 / Details: pH 5.8 | ||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: May 1, 1997 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→30 Å / Num. obs: 34266 / % possible obs: 98.1 % / Observed criterion σ(I): 0 / Biso Wilson estimate: 39.1 Å2 / Rsym value: 0.05 |
| Reflection shell | Resolution: 2.5→2.59 Å / Mean I/σ(I) obs: 2.8 / Rsym value: 0.252 / % possible all: 95 |
| Reflection | *PLUS Rmerge(I) obs: 0.05 |
| Reflection shell | *PLUS % possible obs: 95 % / Rmerge(I) obs: 0.252 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2ACE Resolution: 2.5→30 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.66 Å / Rfactor Rfree error: 0.021 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.286 |
