SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 10-STRANDED BARREL THIS IS REPRESENTED BY A 11-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "BA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 10-STRANDED BARREL THIS IS REPRESENTED BY A 11-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "DA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 7-STRANDED BARREL THIS IS REPRESENTED BY A 8-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "EA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "FA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
温度: 291 K / 手法: マイクロバッチ法 / pH: 6.3 詳細: MICROBATCH METHOD UNDER OIL WAS USED. 15.1 MG/ML OF PROTEIN SOLUTION CONTAINING 0.02M DTT WAS MIXED WITH 1.65M SODIUM ACETATE AND 0.1M MES PH 6.3. THE CRYSTALLIZATION TEMPERATURE WAS 291 K. ...詳細: MICROBATCH METHOD UNDER OIL WAS USED. 15.1 MG/ML OF PROTEIN SOLUTION CONTAINING 0.02M DTT WAS MIXED WITH 1.65M SODIUM ACETATE AND 0.1M MES PH 6.3. THE CRYSTALLIZATION TEMPERATURE WAS 291 K. PARATONE-N OIL MIXED WITH 10% W/W OF GLYCEROL WAS USED FOR CRYOPROTECTION LIGAND BOUND CRYSTALS WERE OBTAINED BY 7 H SOAKING OF NATIVE CRYSTALS IN SOLUTION CONTAINING 1.2M SODIUM ACETATE PH 6.3, 20MM AMMONIUM SULFATE, 2MM MAGNESIUM SULFATE AND 5MM GUANOSINE
プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray
放射波長
波長: 1.5418 Å / 相対比: 1
反射
解像度: 2.4→15 Å / Num. obs: 58580 / % possible obs: 96.9 % / Observed criterion σ(I): -0.5 / 冗長度: 4.99 % / Biso Wilson estimate: 11.4 Å2 / Rmerge(I) obs: 0.118 / Net I/σ(I): 16.9
反射 シェル
解像度: 2.4→2.49 Å / Rmerge(I) obs: 0.332 / Mean I/σ(I) obs: 3.9 / % possible all: 92.7
-
解析
ソフトウェア
名称
バージョン
分類
CNS
1.1
精密化
HKL-2000
データ削減
HKL-2000
データスケーリング
CNS
位相決定
精密化
構造決定の手法: 分子置換 開始モデル: THE REFINED STRUCTURE OF PURINE NUCLEOSIDE PEPTIDASE FROM THERMUS THERMOPHILUS WAS USED AS STARTING MODEL FOR REFINEMENT 解像度: 2.4→14.93 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 253080.12 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 THERMUS THERMOPHILUS (バクテリア)
THERMUS THERMOPHILUS (バクテリア) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード





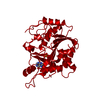



























 PDBj
PDBj 集合体
集合体

 分子量: 96.063 Da / 分子数: 6 / 由来タイプ: 合成 / 式: SO4
分子量: 96.063 Da / 分子数: 6 / 由来タイプ: 合成 / 式: SO4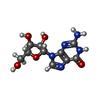
 分子量: 283.241 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C10H13N5O5
分子量: 283.241 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C10H13N5O5 分子量: 18.015 Da / 分子数: 258 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 258 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析