 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1odi | ||||||
|---|---|---|---|---|---|---|---|
| Title | Purine nucleoside phosphorylase from Thermus Thermophilus | ||||||
 Components Components | PURINE NUCLEOSIDE PHOSPHORYLASE | ||||||
 Keywords Keywords | TRANSFERASE / NUCLEOSIDE PHOSPHORYLASE / ALPHA-BETA PROTEIN / ADENOSINE / RIKEN STRUCTURAL GENOMICS/PROTEOMICS INITIATIVE / RSGI / STRUCTURAL GENOMICS | ||||||
| Function / homology |  Function and homology information Function and homology informationuridine phosphorylase / nucleoside catabolic process / uridine phosphorylase activity / cytosol Similarity search - Function | ||||||
| Biological species |   THERMUS THERMOPHILUS (bacteria) THERMUS THERMOPHILUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 2.4 Å X-RAY DIFFRACTION / OTHER / Resolution: 2.4 Å | ||||||
 Authors Authors | Tahirov, T.H. / Inagaki, E. / Miyano, M. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2004 Title: Crystal Structure of Purine Nucleoside Phosphorylase from Thermus Thermophilus Authors: Tahirov, T.H. / Inagaki, E. / Ohshima, N. / Kitao, T. / Kuroishi, C. / Ukita, Y. / Takio, K. / Kobayashi, M. / Kuramitsu, S. / Yokoyama, S. / Miyano, M. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA","BA" IN EACH CHAIN ON SHEET RECORDS ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA","BA" IN EACH CHAIN ON SHEET RECORDS BELOW ARE ACTUALLY 10-STRANDED BARRELS THIS IS REPRESENTED BY A 11-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "CA", "DA", "EA", "FA" IN EACH CHAIN ON SHEET RECORDS BELOW ARE ACTUALLY AN 8-STRANDED BARRELS THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1odi.cif.gz | 281.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1odi.ent.gz | 229 KB | Display | PDB format |
| PDBx/mmJSON format | 1odi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/od/1odiftp://data.pdbj.org/pub/pdb/validation_reports/od/1odi | HTTPS FTP |
|---|
-Related structure data

































-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 25444.100 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) THERMUS THERMOPHILUS (bacteria) / Strain: HB8 / Production host: References: UniProt: Q5SID9*PLUS, S-methyl-5'-thioadenosine phosphorylase #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-ADN / 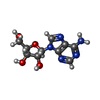  Mass: 267.241 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H13N5O4 Mass: 267.241 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C10H13N5O4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 391 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 391 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 49.9 % Description: THE REFINED STRUCTURE OF PURINE NUCLEOSIDE PEPTIDASE FROM THERMUS THERMOPHILUS WAS USED AS STARTING MODEL FOR REFINEMENT | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: microbatch / pH: 6.3 Details: MICROBATCH METHOD UNDER OIL WAS USED. 15.1 MG/ML OF PROTEIN SOLUTION CONTAINING 0.02M DTT WAS MIXED WITH 1.65M SODIUM ACETATE AND 0.1M MES PH 6.3. THE CRYSTALLIZATION TEMPERATURE WAS 291 K. ...Details: MICROBATCH METHOD UNDER OIL WAS USED. 15.1 MG/ML OF PROTEIN SOLUTION CONTAINING 0.02M DTT WAS MIXED WITH 1.65M SODIUM ACETATE AND 0.1M MES PH 6.3. THE CRYSTALLIZATION TEMPERATURE WAS 291 K. PARATONE-N OIL MIXED WITH 10% W/W OF GLYCEROL WAS USED FOR CRYOPROTECTION. LIGAND BOUND CRYSTALS WERE OBTAINED BY 7 H SOAKING OF NATIVE CRYSTALS IN SOLUTION CONTAINING 1.2M SODIUM ACETATE PH 6.3, 20MM AMMONIUM SULFATE, 2MM MAGNESIUM SULFATE AND 5MM ADENOSINE | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 291 K / Method: microseeding | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-D / Wavelength: 1.5418 |
| Detector | Type: RIGAKU IMAGE PLATE, RAXIS-VII / Detector: IMAGE PLATE / Date: Jan 15, 2003 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→30 Å / Num. obs: 56385 / % possible obs: 94.4 % / Observed criterion σ(I): -0.5 / Redundancy: 4.1 % / Biso Wilson estimate: 14.8 Å2 / Rmerge(I) obs: 0.088 / Net I/σ(I): 13.2 |
| Reflection shell | Resolution: 2.4→2.49 Å / Rmerge(I) obs: 0.335 / Mean I/σ(I) obs: 2.9 / % possible all: 88.8 |
| Reflection shell | *PLUS % possible obs: 88.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER / Resolution: 2.4→19.96 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 247589.73 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 31.0905 Å2 / ksol: 0.350542 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→19.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
