 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1odi | ||||||
|---|---|---|---|---|---|---|---|
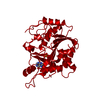
| タイトル | Purine nucleoside phosphorylase from Thermus Thermophilus | ||||||
 要素 要素 | PURINE NUCLEOSIDE PHOSPHORYLASE | ||||||
 キーワード キーワード | TRANSFERASE (転移酵素) / NUCLEOSIDE PHOSPHORYLASE / ALPHA-BETA PROTEIN / ADENOSINE (アデノシン) / RIKEN STRUCTURAL GENOMICS/PROTEOMICS INITIATIVE / RSGI / STRUCTURAL GENOMICS (構造ゲノミクス) | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報 | ||||||
| 生物種 |   THERMUS THERMOPHILUS (サーマス・サーモフィルス) THERMUS THERMOPHILUS (サーマス・サーモフィルス) | ||||||
| 手法 | X線回折 / その他 / 解像度: 2.4 Å | ||||||
 データ登録者 データ登録者 | Tahirov, T.H. / Inagaki, E. / Miyano, M. | ||||||
 引用 引用 | ジャーナル: J.Mol.Biol. / 年: 2004 タイトル: Crystal Structure of Purine Nucleoside Phosphorylase from Thermus Thermophilus 著者: Tahirov, T.H. / Inagaki, E. / Ohshima, N. / Kitao, T. / Kuroishi, C. / Ukita, Y. / Takio, K. / Kobayashi, M. / Kuramitsu, S. / Yokoyama, S. / Miyano, M. | ||||||
| 履歴 |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA","BA" IN EACH CHAIN ON SHEET RECORDS ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA","BA" IN EACH CHAIN ON SHEET RECORDS BELOW ARE ACTUALLY 10-STRANDED BARRELS THIS IS REPRESENTED BY A 11-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "CA", "DA", "EA", "FA" IN EACH CHAIN ON SHEET RECORDS BELOW ARE ACTUALLY AN 8-STRANDED BARRELS THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. |
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1odi.cif.gz | 277.5 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1odi.ent.gz | 232.6 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1odi.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/od/1odiftp://data.pdbj.org/pub/pdb/validation_reports/od/1odi | HTTPS FTP |
|---|
-関連構造データ

































-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 25444.100 Da / 分子数: 6 / 由来タイプ: 組換発現 由来: (組換発現) THERMUS THERMOPHILUS (サーマス・サーモフィルス)株: HB8 / 発現宿主: ESCHERICHIA COLI BL21(DE3) (大腸菌)参照: UniProt: Q5SID9*PLUS, S-methyl-5'-thioadenosine phosphorylase#2: 化合物 | ChemComp-SO4 / 硫酸塩  分子量: 96.063 Da / 分子数: 8 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 8 / 由来タイプ: 合成 / 式: SO4#3: 化合物 | ChemComp-ADN / アデノシン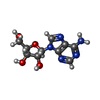  分子量: 267.241 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C10H13N5O4 分子量: 267.241 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C10H13N5O4#4: 水 | ChemComp-HOH / | 水 分子量: 18.015 Da / 分子数: 391 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 391 / 由来タイプ: 天然 / 式: H2O |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.5 Å3/Da / 溶媒含有率: 49.9 % 解説: THE REFINED STRUCTURE OF PURINE NUCLEOSIDE PEPTIDASE FROM THERMUS THERMOPHILUS WAS USED AS STARTING MODEL FOR REFINEMENT | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 291 K / 手法: マイクロバッチ法 / pH: 6.3 詳細: MICROBATCH METHOD UNDER OIL WAS USED. 15.1 MG/ML OF PROTEIN SOLUTION CONTAINING 0.02M DTT WAS MIXED WITH 1.65M SODIUM ACETATE AND 0.1M MES PH 6.3. THE CRYSTALLIZATION TEMPERATURE WAS 291 K. ...詳細: MICROBATCH METHOD UNDER OIL WAS USED. 15.1 MG/ML OF PROTEIN SOLUTION CONTAINING 0.02M DTT WAS MIXED WITH 1.65M SODIUM ACETATE AND 0.1M MES PH 6.3. THE CRYSTALLIZATION TEMPERATURE WAS 291 K. PARATONE-N OIL MIXED WITH 10% W/W OF GLYCEROL WAS USED FOR CRYOPROTECTION. LIGAND BOUND CRYSTALS WERE OBTAINED BY 7 H SOAKING OF NATIVE CRYSTALS IN SOLUTION CONTAINING 1.2M SODIUM ACETATE PH 6.3, 20MM AMMONIUM SULFATE, 2MM MAGNESIUM SULFATE AND 5MM ADENOSINE | ||||||||||||||||||||||||||||||
| 結晶化 | *PLUS 温度: 291 K / 手法: microseeding | ||||||||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: 回転陽極 / タイプ: RIGAKU FR-D / 波長: 1.5418 |
| 検出器 | タイプ: RIGAKU IMAGE PLATE, RAXIS-VII / 検出器: IMAGE PLATE / 日付: 2003年1月15日 / 詳細: MIRRORS |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.5418 Å / 相対比: 1 |
| 反射 | 解像度: 2.4→30 Å / Num. obs: 56385 / % possible obs: 94.4 % / Observed criterion σ(I): -0.5 / 冗長度: 4.1 % / Biso Wilson estimate: 14.8 Å2 / Rmerge(I) obs: 0.088 / Net I/σ(I): 13.2 |
| 反射 シェル | 解像度: 2.4→2.49 Å / Rmerge(I) obs: 0.335 / Mean I/σ(I) obs: 2.9 / % possible all: 88.8 |
| 反射 シェル | *PLUS % possible obs: 88.8 % |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: その他 / 解像度: 2.4→19.96 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 247589.73 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 31.0905 Å2 / ksol: 0.350542 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 27.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2.4→19.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2.4→2.55 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: CNS / バージョン: 1.1 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
