coagulation factor Xa / Defective factor IX causes thrombophilia / Defective cofactor function of FVIIIa variant / Defective F9 variant does not activate FX / : / positive regulation of TOR signaling / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / : / Gamma-carboxylation of protein precursors / Removal of aminoterminal propeptides from gamma-carboxylated proteins ...coagulation factor Xa / Defective factor IX causes thrombophilia / Defective cofactor function of FVIIIa variant / Defective F9 variant does not activate FX / : / positive regulation of TOR signaling / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / : / Gamma-carboxylation of protein precursors / Removal of aminoterminal propeptides from gamma-carboxylated proteins / : / phospholipid binding / Golgi lumen / blood coagulation / positive regulation of cell migration / endoplasmic reticulum lumen / serine-type endopeptidase activity / external side of plasma membrane / calcium ion binding / proteolysis / : / extracellular region / plasma membrane Similarity search - Function
SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 6-STRANDED BARREL THIS IS REPRESENTED BY A 7-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
ACTIVATEDFACTORXAHEAVYCHAIN / COAGULATION FACTOR X / STUART FACTOR / STUART-PROWER FACTOR
Mass: 28550.596 Da / Num. of mol.: 1 / Source method: isolated from a natural source Details: PURCHASED FROM ENZYME RESEARCH LABS. ISOLATED FROM HUMAN BLOOD Source: (natural) HOMO SAPIENS (human) / References: UniProt: P00742, coagulation factor Xa
#2: Protein
FACTORXLIGHTCHAIN / COAGULATION FACTOR X / STUART FACTOR / STUART-PROWER FACTOR
Mass: 15210.793 Da / Num. of mol.: 1 / Fragment: ACTIVATED DESGLA, RESIDUES 46-179 / Source method: isolated from a natural source Details: PURCHASED FROM ENZYME RESEARCH LABS. ISOLATED FROM HUMAN BLOOD Source: (natural) HOMO SAPIENS (human) / References: UniProt: P00742, coagulation factor Xa
Mass: 18.015 Da / Num. of mol.: 272 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has protein modification
Y
Sequence details
SOME RESIDUES IN CHAIN ARE NOT SEEN IN THE ELECTRON DENSITY. SEQUENCE DATABASE RESIDUES 1-45 (THE ...SOME RESIDUES IN CHAIN ARE NOT SEEN IN THE ELECTRON DENSITY. SEQUENCE DATABASE RESIDUES 1-45 (THE GLA DOMAIN) WERE BIOCHEMICALLY REMOVED IN CHAIN B
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 1.81 Å3/Da / Density % sol: 49.3 % / Description: NONE
Crystal grow
Method: vapor diffusion / pH: 5.75 Details: CRYSTALLISATION WAS CARRIED OUT USING VAPOUR DIFFUSION IN 2UL DROPS CONTAINING A 1:1 MIXTURE OF PROTEIN AND WELL SOLUTION. WELL SOLUTION CONTAINED 16-20% PEG 6K, 50 MM MES-NAOH (PH 5.5-6), 5 ...Details: CRYSTALLISATION WAS CARRIED OUT USING VAPOUR DIFFUSION IN 2UL DROPS CONTAINING A 1:1 MIXTURE OF PROTEIN AND WELL SOLUTION. WELL SOLUTION CONTAINED 16-20% PEG 6K, 50 MM MES-NAOH (PH 5.5-6), 5 MM CACL2 AND 50 MM NACL.
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.961 Å / Relative weight: 1
Reflection
Resolution: 1.69→44.54 Å / Num. obs: 35816 / % possible obs: 97.6 % / Observed criterion σ(I): 0 / Redundancy: 5.5 % / Biso Wilson estimate: 20.615 Å2 / Rmerge(I) obs: 0.08 / Net I/σ(I): 14.4
Reflection shell
Resolution: 1.69→1.78 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.48 / Mean I/σ(I) obs: 3.4 / % possible all: 100
-
Processing
Software
Name
Version
Classification
REFMAC
5.5.0109
refinement
MOSFLM
datareduction
SCALA
datascaling
Refinement
Method to determine structure: OTHER Starting model: NONE Resolution: 1.7→20 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.935 / SU B: 2.119 / SU ML: 0.07 / Cross valid method: THROUGHOUT / ESU R: 0.106 / ESU R Free: 0.107 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.22473
1753
5 %
RANDOM
Rwork
0.1861
-
-
-
obs
0.18801
33357
100 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

























































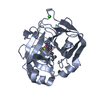







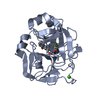









































 PDBj
PDBj







 Assembly
Assembly
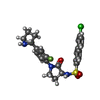



 Mass: 487.974 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H23ClFN3O3S
Mass: 487.974 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H23ClFN3O3S Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.961
/ Beamline: ID29 / Wavelength: 0.961  Processing
Processing