 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1v3x: Factor Xa in complex with the inhibitor 1-[6-methyl-4,5,6,7-tetra... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1v3x | ||||||
|---|---|---|---|---|---|---|---|
| Title | Factor Xa in complex with the inhibitor 1-[6-methyl-4,5,6,7-tetrahydrothiazolo(5,4-c)pyridin-2-yl] carbonyl-2-carbamoyl-4-(6-chloronaphth-2-ylsulphonyl)piperazine | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / glycoprotein / serine protease / plasma / blood coagulation factor / protein inhibitor complex / calcium-binding | ||||||
| Function / homology |  Function and homology information Function and homology informationcoagulation factor Xa / Defective factor IX causes thrombophilia / Defective cofactor function of FVIIIa variant / Defective F9 variant does not activate FX / : / positive regulation of TOR signaling / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / : / Gamma-carboxylation of protein precursors / Removal of aminoterminal propeptides from gamma-carboxylated proteins ...coagulation factor Xa / Defective factor IX causes thrombophilia / Defective cofactor function of FVIIIa variant / Defective F9 variant does not activate FX / : / positive regulation of TOR signaling / Transport of gamma-carboxylated protein precursors from the endoplasmic reticulum to the Golgi apparatus / : / Gamma-carboxylation of protein precursors / Removal of aminoterminal propeptides from gamma-carboxylated proteins / : / phospholipid binding / Golgi lumen / blood coagulation / positive regulation of cell migration / endoplasmic reticulum lumen / serine-type endopeptidase activity / external side of plasma membrane / calcium ion binding / proteolysis / : / extracellular region / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / RIGID BODY REFINEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / RIGID BODY REFINEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Suzuki, M. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2004 Title: Synthesis and conformational analysis of a non-amidine factor Xa inhibitor that incorporates 5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine as S4 binding element Authors: Haginoya, N. / Kobayashi, S. / Komoriya, S. / Yoshino, T. / Suzuki, M. / Shimada, T. / Watanabe, K. / Hirokawa, Y. / Furugori, T. / Nagahara, T. #1: Journal: Proc.Natl.Acad.Sci.Usa / Year: 1998Title: Structural basis for chemical inhibition of human blood coagulation factor Xa Authors: Kamata, K. / Kawamoto, H. / Honma, T. / Iwama, T. / Kim, S.H. #2: Journal: J.Biol.Chem. / Year: 1996Title: X-ray structure of active site-inhibited clotting factor Xa. Implications for drug design and substrate recognition Authors: Brandstetter, H. / Kuhne, A. / Bode, W. / Huber, R. / von der Saal, W. / Wirthensohn, K. / Engh, R.A. #3: Journal: J.Mol.Biol. / Year: 1993Title: Structure of human des(1-45) factor Xa at 2.2 A resolution Authors: Padmanabhan, K. / Padmanabhan, K.P. / Tulinsky, A. / Park, C.H. / Bode, W. / Huber, R. / Blankenship, D.T. / Cardin, A.D. / Kisiel, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1v3x.cif.gz | 75.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1v3x.ent.gz | 54.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1v3x.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/v3/1v3xftp://data.pdbj.org/pub/pdb/validation_reports/v3/1v3x | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1faxS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 26346.000 Da / Num. of mol.: 1 / Fragment: Residues 16-243 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / References: UniProt: P00742, coagulation factor Xa | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein | Mass: 5589.234 Da / Num. of mol.: 1 / Fragment: Residues 87-138 / Source method: isolated from a natural source / Details: proteolytic cleavage product / Source: (natural) Homo sapiens (human) / References: UniProt: P00742, coagulation factor Xa | ||||||
| #3: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-D76 / ( | 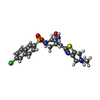  Mass: 534.051 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H24ClN5O4S2 Mass: 534.051 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H24ClN5O4S2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 161 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 161 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.49 Å3/Da / Density % sol: 50.54 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, macro-seeding, soaking / pH: 5 Details: PEG6000, sodium acetate, Malate imidazole, Calcium chloride, pH 5.00, vapor diffusion, macro-seeding, Soaking, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.542 / Wavelength: 1.5418 Å | |||||||||
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Dec 3, 1998 | |||||||||
| Radiation | Monochromator: GRAPHITE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 2.2→53.124 Å / Num. all: 15999 / Num. obs: 15999 / % possible obs: 96 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Biso Wilson estimate: 35.621 Å2 / Rmerge(I) obs: 0.044 / Rsym value: 0.044 / Net I/σ(I): 13.6 | |||||||||
| Reflection shell | Resolution: 2.2→2.27 Å / Redundancy: 2.6 % / Rmerge(I) obs: 0.259 / Mean I/σ(I) obs: 2.9 / Num. unique all: 1332 / Rsym value: 0.259 / % possible all: 90.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: RIGID BODY REFINEMENT Starting model: 1FAX Resolution: 2.2→25 Å / Cor.coef. Fo:Fc: 0.931 / Cor.coef. Fo:Fc free: 0.921 / SU B: 5.151 / SU ML: 0.135 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.271 / ESU R Free: 0.204 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: BABINET MODEL PARAMETERS FOR MASK CALCULATION | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.912 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→25 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.277 Å / Total num. of bins used: 15 /
|
