 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2x3f: Crystal Structure of the Methicillin-Resistant Staphylococcus aur... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2x3f | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Methicillin-Resistant Staphylococcus aureus Sar2676, a Pantothenate Synthetase. | ||||||
 Components Components | PANTHOTHENATE SYNTHETASE | ||||||
 Keywords Keywords | LIGASE / ATP-BINDING / NUCLEOTIDE-BINDING / PANTOTHENATE BIOSYNTHESIS | ||||||
| Function / homology |  Function and homology information Function and homology informationpantoate-beta-alanine ligase (AMP-forming) / pantoate-beta-alanine ligase activity / pantothenate biosynthetic process / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |   STAPHYLOCOCCUS AUREUS SUBSP. AUREUS (bacteria) STAPHYLOCOCCUS AUREUS SUBSP. AUREUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | ||||||
 Authors Authors | Oke, M. / Carter, L.G. / Johnson, K.A. / Liu, H. / Mcmahon, S.A. / White, M.F. / Naismith, J.H. | ||||||
 Citation Citation | Journal: J.Struct.Funct.Genom. / Year: 2010 Title: The Scottish Structural Proteomics Facility: Targets, Methods and Outputs. Authors: Oke, M. / Carter, L.G. / Johnson, K.A. / Liu, H. / Mcmahon, S.A. / Yan, X. / Kerou, M. / Weikart, N.D. / Kadi, N. / Sheikh, M.A. / Schmelz, S. / Dorward, M. / Zawadzki, M. / Cozens, C. / ...Authors: Oke, M. / Carter, L.G. / Johnson, K.A. / Liu, H. / Mcmahon, S.A. / Yan, X. / Kerou, M. / Weikart, N.D. / Kadi, N. / Sheikh, M.A. / Schmelz, S. / Dorward, M. / Zawadzki, M. / Cozens, C. / Falconer, H. / Powers, H. / Overton, I.M. / Van Niekerk, C.A.J. / Peng, X. / Patel, P. / Garrett, R.A. / Prangishvili, D. / Botting, C.H. / Coote, P.J. / Dryden, D.T.F. / Barton, G.J. / Schwarz-Linek, U. / Challis, G.L. / Taylor, G.L. / White, M.F. / Naismith, J.H. #1: Journal: Acta Crystallogr.,Sect.F / Year: 2007 Title: Purification, Crystallization and Data Collection of Methicillin-Resistant Staphylococcus Aureus Sar2676, a Pantothenate Synthetase. Authors: Seetharamappa, J. / Oke, M. / Liu, H. / Mcmahon, S.A. / Johnson, K.A. / Carter, L. / Dorward, M. / Zawadzki, M. / Overton, I.M. / Van Niekirk, C.A.J. / Graham, S. / Botting, C.H. / Taylor, G. ...Authors: Seetharamappa, J. / Oke, M. / Liu, H. / Mcmahon, S.A. / Johnson, K.A. / Carter, L. / Dorward, M. / Zawadzki, M. / Overton, I.M. / Van Niekirk, C.A.J. / Graham, S. / Botting, C.H. / Taylor, G.L. / White, M.F. / Barton, G.J. / Coote, P.J. / Naismith, J.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2x3f.cif.gz | 234.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2x3f.ent.gz | 190.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2x3f.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/x3/2x3fftp://data.pdbj.org/pub/pdb/validation_reports/x3/2x3f | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 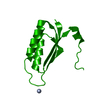 2ivyC  2jg5C  2jg6C  2vw8C  2vxzC  2wj9C  2x0oC  2x3dC  2x3eC  2x3gC  2x3lC  2x3mC  2x3nC  2x3oC  2x48C  2x4gC  2x4hC  2x4iC  2x4jC  2x4kC  2x4lC  2x5cC  2x5dC  2x5fC  2x5gC  2x5hC  2x5pC  2x5qC  2x5rC  2x5tC  2x7bC  2x7iC  2xu2C  1v8fS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31584.105 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) STAPHYLOCOCCUS AUREUS SUBSP. AUREUS (bacteria)Strain: MRSA252 / Plasmid: PDEST14 / Production host: References: UniProt: Q6GDK5, pantoate-beta-alanine ligase (AMP-forming) #2: Chemical | 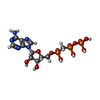  Mass: 505.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-CPP, energy-carrying molecule analogue*YM#3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 45 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.5 Details: 10% PEG 8000, 0.1M NA MES PH6.5, 0.2M ZN ACETATE WITH 11.5MGML-1 AMP-CPP. THE CRYSTALS WERE CRYOPROTECTED WITH OIL. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 / Beamline: ID14-2 / Wavelength: 0.933 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Oct 11, 2007 / Details: TOROIDAL ZERODUR MIRROR |
| Radiation | Monochromator: DIAMOND (111), GE(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→28.59 Å / Num. obs: 39073 / % possible obs: 99.6 % / Observed criterion σ(I): 0 / Redundancy: 11.2 % / Rmerge(I) obs: 0.05 / Net I/σ(I): 14.5 |
| Reflection shell | Resolution: 1.95→2 Å / Redundancy: 10.4 % / Rmerge(I) obs: 0.28 / Mean I/σ(I) obs: 2 / % possible all: 98 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1V8F Resolution: 1.95→28.59 Å / Cor.coef. Fo:Fc: 0.94 / Cor.coef. Fo:Fc free: 0.926 / SU B: 8.227 / SU ML: 0.109 / Cross valid method: THROUGHOUT / ESU R: 0.192 / ESU R Free: 0.164 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS THE STRUCTURE IS ONLY ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS THE STRUCTURE IS ONLY ORDERED FROM RESIDUE 2 TO 282
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.115 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→28.59 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|