 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-31916 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | 2.56 A structure of influenza hemagglutinin (HA) trimer | ||||||||||||||||||
 Map data Map data | |||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
 Keywords Keywords | Influenza hemagglutinin (HA) trimer / VIRAL PROTEIN | ||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationviral budding from plasma membrane / clathrin-dependent endocytosis of virus by host cell / host cell surface receptor binding / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / virion attachment to host cell / host cell plasma membrane / virion membrane / membrane Similarity search - Function | ||||||||||||||||||
| Biological species |  H3N2 subtype (virus) / Influenza A virus (strain A/Hong Kong/1/1968 H3N2) H3N2 subtype (virus) / Influenza A virus (strain A/Hong Kong/1/1968 H3N2) | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.56 Å | ||||||||||||||||||
 Authors Authors | Fan HC / Zhang Y | ||||||||||||||||||
| Funding support |  China, 5 items China, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: Nat Commun / Year: 2021 Title: A cryo-electron microscopy support film formed by 2D crystals of hydrophobin HFBI. Authors: Hongcheng Fan / Bo Wang / Yan Zhang / Yun Zhu / Bo Song / Haijin Xu / Yujia Zhai / Mingqiang Qiao / Fei Sun / Abstract: Cryo-electron microscopy (cryo-EM) has become a powerful tool to resolve high-resolution structures of biomacromolecules in solution. However, air-water interface induced preferred orientations, ...Cryo-electron microscopy (cryo-EM) has become a powerful tool to resolve high-resolution structures of biomacromolecules in solution. However, air-water interface induced preferred orientations, dissociation or denaturation of biomacromolecules during cryo-vitrification remains a limiting factor for many specimens. To solve this bottleneck, we developed a cryo-EM support film using 2D crystals of hydrophobin HFBI. The hydrophilic side of the HFBI film adsorbs protein particles via electrostatic interactions and sequesters them from the air-water interface, allowing the formation of sufficiently thin ice for high-quality data collection. The particle orientation distribution can be regulated by adjusting the buffer pH. Using this support, we determined the cryo-EM structures of catalase (2.29 Å) and influenza haemagglutinin trimer (2.56 Å), which exhibited strong preferred orientations using a conventional cryo-vitrification protocol. We further show that the HFBI film is suitable to obtain high-resolution structures of small proteins, including aldolase (150 kDa, 3.28 Å) and haemoglobin (64 kDa, 3.6 Å). Our work suggests that HFBI films may have broad future applications in increasing the success rate and efficiency of cryo-EM. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_31916.map.gz | 117.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-31916-v30.xmlemd-31916.xml | 19.4 KB 19.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_31916_fsc.xml | 11.1 KB | Display | FSC data file |
| Images |  emd_31916.png emd_31916.png | 93.1 KB | ||
| Filedesc metadata | emd-31916.cif.gz | 6.5 KB | ||
| Others | emd_31916_half_map_1.map.gzemd_31916_half_map_2.map.gz | 116.1 MB 116.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-31916ftp://ftp.pdbj.org/pub/emdb/structures/EMD-31916 http://ftp.pdbj.org/pub/emdb/structures/EMD-31916ftp://ftp.pdbj.org/pub/emdb/structures/EMD-31916 | HTTPS FTP |
-Related structure data
| Related structure data |  7vdfMC  7vd8C  7vd9C  7vdaC  7vdcC  7vdeC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |













-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |






-Map
| File | Download / File: emd_31916.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.76 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: #1
| File | emd_31916_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Half map: #2
| File | emd_31916_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






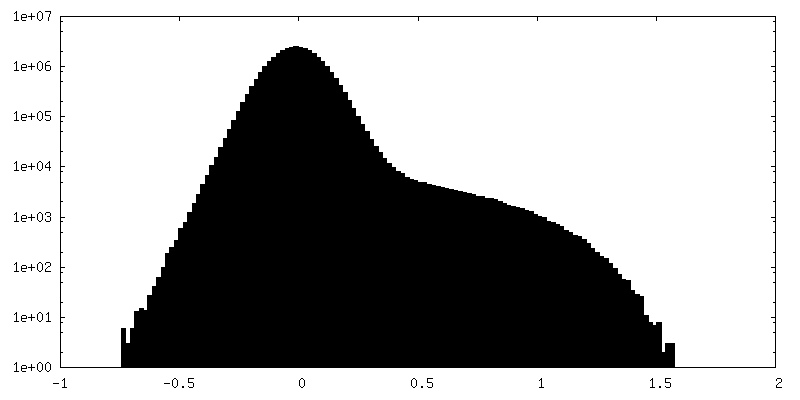
- Sample components
Sample components
-Entire : Influenza hemagglutinin (HA) trimer
| Entire | Name: Influenza hemagglutinin (HA) trimer |
|---|---|
| Components |
|
-Supramolecule #1: Influenza hemagglutinin (HA) trimer
| Supramolecule | Name: Influenza hemagglutinin (HA) trimer / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: H3N2 subtype (virus) |
| Molecular weight | Theoretical: 190 KDa |
-Macromolecule #1: Hemagglutinin
| Macromolecule | Name: Hemagglutinin / type: protein_or_peptide / ID: 1 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Influenza A virus (strain A/Hong Kong/1/1968 H3N2) Strain: A/Hong Kong/1/1968 H3N2 |
| Molecular weight | Theoretical: 62.855215 KDa |
| Recombinant expression | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Sequence | String: QDLPGNDNST ATLCLGHHAV PNGTLVKTIT DDQIEVTNAT ELVQSSSTGK ICNNPHRILD GIDCTLIDAL LGDPHCDVFQ NETWDLFVE RSKAFSNCYP YDVPDYASLR SLVASSGTLE FITEGFTWTG VTQNGGSNAC KRGPGSGFFS RLNWLTKSGS T YPVLNVTM ...String: QDLPGNDNST ATLCLGHHAV PNGTLVKTIT DDQIEVTNAT ELVQSSSTGK ICNNPHRILD GIDCTLIDAL LGDPHCDVFQ NETWDLFVE RSKAFSNCYP YDVPDYASLR SLVASSGTLE FITEGFTWTG VTQNGGSNAC KRGPGSGFFS RLNWLTKSGS T YPVLNVTM PNNDNFDKLY IWGVHHPSTN QEQTSLYVQA SGRVTVSTRR SQQTIIPNIG SRPWVRGLSS RISIYWTIVK PG DVLVINS NGNLIAPRGY FKMRTGKSSI MRSDAPIDTC ISECITPNGS IPNDKPFQNV NKITYGACPK YVKQNTLKLA TGM RNVPEK QTRGLFGAIA GFIENGWEGM IDGWYGFRHQ NSEGTGQAAD LKSTQAAIDQ INGKLNRVIE KTNEKFHQIE KEFS EVEGR IQDLEKYVED TKIDLWSYNA ELLVALENQH TIDLTDSEMN KLFEKTRRQL RENAEDMGNG CFKIYHKCDN ACIES IRNG TYDHDVYRDE ALNNRFQIKG VELKSGYDGG GGTGGGGTGR MKQIEDKIEE ILSKIYHIEN EIARIKKLIG ERHHHH HH UniProtKB: Hemagglutinin |
-Macromolecule #2: 2-acetamido-2-deoxy-beta-D-glucopyranose
| Macromolecule | Name: 2-acetamido-2-deoxy-beta-D-glucopyranose / type: ligand / ID: 2 / Number of copies: 12 / Formula: NAG |
|---|---|
| Molecular weight | Theoretical: 221.208 Da |
| Chemical component information |  ChemComp-NAG: |
-Macromolecule #3: beta-D-mannopyranose
| Macromolecule | Name: beta-D-mannopyranose / type: ligand / ID: 3 / Number of copies: 3 / Formula: BMA |
|---|---|
| Molecular weight | Theoretical: 180.156 Da |
| Chemical component information |  ChemComp-BMA: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Grid | Material: NICKEL/TITANIUM / Mesh: 300 |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Image recording | Film or detector model: GATAN K2 QUANTUM (4k x 4k) / Detector mode: SUPER-RESOLUTION / Number real images: 4574 / Average electron dose: 60.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal magnification: 165000 |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |

