Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-0152: RELION-3.0 reconstruction of influenza hemagglutinin (HA) trimer ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-0152 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | RELION-3.0 reconstruction of influenza hemagglutinin (HA) trimer using particles from micrographs tilted at 40 degrees in EMPIAR-10097 | |||||||||
 Map data Map data | postprocessed map | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
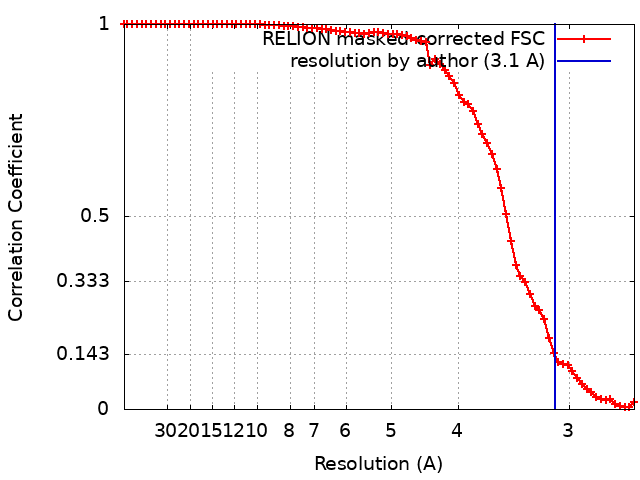
| Method | single particle reconstruction / cryo EM / Resolution: 3.1 Å | |||||||||
 Authors Authors | Zivanov J / Nakane T / Scheres SHW | |||||||||
| Funding support |  United Kingdom, 1 items United Kingdom, 1 items
| |||||||||
 Citation Citation | Journal: Nat Methods / Year: 2017 Title: Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Authors: Yong Zi Tan / Philip R Baldwin / Joseph H Davis / James R Williamson / Clinton S Potter / Bridget Carragher / Dmitry Lyumkis /  Abstract: We present a strategy for tackling preferred specimen orientation in single-particle cryogenic electron microscopy by employing tilts during data collection. We also describe a tool to quantify the ...We present a strategy for tackling preferred specimen orientation in single-particle cryogenic electron microscopy by employing tilts during data collection. We also describe a tool to quantify the resulting directional resolution using 3D Fourier shell correlation volumes. We applied these methods to determine the structures at near-atomic resolution of the influenza hemagglutinin trimer, which adopts a highly preferred specimen orientation, and of ribosomal biogenesis intermediates, which adopt moderately preferred orientations. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
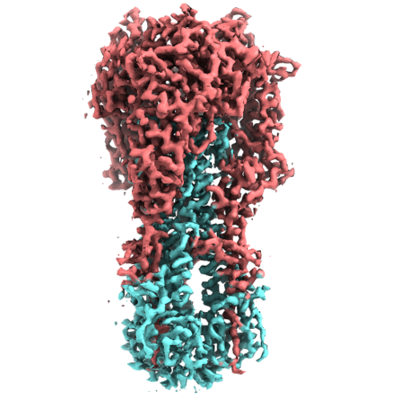
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0152.map.gz | 35.8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0152-v30.xmlemd-0152.xml | 19.1 KB 19.1 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_0152_fsc.xml | 7.7 KB | Display | FSC data file |
| Images |  emd_0152.png emd_0152.png | 143.9 KB | ||
| Masks | emd_0152_msk_1.map | 38.4 MB | Mask map | |
| Others | emd_0152_half_map_1.map.gzemd_0152_half_map_2.map.gz | 29.7 MB 29.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0152ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0152 http://ftp.pdbj.org/pub/emdb/structures/EMD-0152ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0152 | HTTPS FTP |
-Related structure data












-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0152.map.gz / Format: CCP4 / Size: 38.4 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | postprocessed map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.31 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_0152_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: unfiltered half map 1
| File | emd_0152_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | unfiltered half map 1 | ||||||||||||
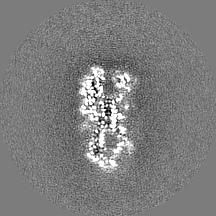
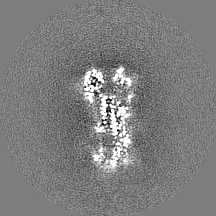
| Projections & Slices |
| ||||||||||||
| Density Histograms |





-Half map: unfiltered half map 2
| File | emd_0152_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | unfiltered half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




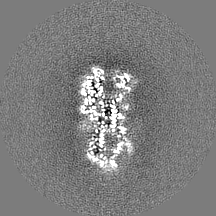
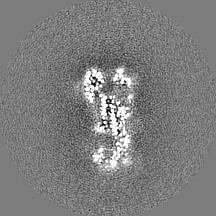

- Sample components
Sample components
-Entire : Influenza hemagglutinin (HA) trimer
| Entire | Name: Influenza hemagglutinin (HA) trimer |
|---|---|
| Components |
|
-Supramolecule #1: Influenza hemagglutinin (HA) trimer
| Supramolecule | Name: Influenza hemagglutinin (HA) trimer / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 150 KDa |
-Macromolecule #1: Influenza hemagglutinin HA1 chain A/Hong Kong/1/1968 H3N2
| Macromolecule | Name: Influenza hemagglutinin HA1 chain A/Hong Kong/1/1968 H3N2 type: protein_or_peptide / ID: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / Strain: HEK293 |
| Sequence | String: ADPGATLCLG HHAVPNGTLV KTITDDQIEV TNATELVQSS STGKICNNPH RILDGIDCTL IDALLGDPHC DVFQNETWDL FVERSKAFS NCYPYDVPDY ASLRSLVASS GTLEFITEGF TWTGVTQNGG SNACKRGPGS GFFSRLNWLT KSGSTYPVLN V TMPNNDNF ...String: ADPGATLCLG HHAVPNGTLV KTITDDQIEV TNATELVQSS STGKICNNPH RILDGIDCTL IDALLGDPHC DVFQNETWDL FVERSKAFS NCYPYDVPDY ASLRSLVASS GTLEFITEGF TWTGVTQNGG SNACKRGPGS GFFSRLNWLT KSGSTYPVLN V TMPNNDNF DKLYIWGVHH PSTNQEQTSL YVQASGRVTV STRRSQQTII PNIGSRPWVR GLSSRISIYW TIVKPGDVLV IN SNGNLIA PRGYFKMRTG KSSIMRSDAP IDTCISECIT PNGSIPNDKP FQNVNKITYG ACPKYVKQNT LKLATGMRNV PEK QTR |
-Macromolecule #2: Influenza hemagglutinin HA2 chain A/Hong Kong/1/1968 H3N2
| Macromolecule | Name: Influenza hemagglutinin HA2 chain A/Hong Kong/1/1968 H3N2 type: protein_or_peptide / ID: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / Strain: HEK293 |
| Sequence | String: GLFGAIAGF IENGWEGMID GWYGFRHQNS EGTGQAADLK STQAAIDQIN GKLNRVIEKT NEKFHQIEKE FSEVEGRIQD L EKYVEDTK IDLWSYNAEL LVALENQHTI DLTDSEMNKL FEKTGRQLRE NAEDMGNGCF KIYHKCDNAC IESIRNGTYD HD VYRDEAL NNRFQIK |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.75 mg/mL |
|---|---|
| Buffer | pH: 7.4 / Details: PBS Buffer |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 80 % / Instrument: GATAN CRYOPLUNGE 3 Details: 3 microliters of 0.75 mg/mL sample was added to a plasma-cleaned (Gatan Solarus) 1.2-micron-hole, 1.3-micron-spacing holey gold grid (made in-house) and plunge-frozen in liquid ethane using ...Details: 3 microliters of 0.75 mg/mL sample was added to a plasma-cleaned (Gatan Solarus) 1.2-micron-hole, 1.3-micron-spacing holey gold grid (made in-house) and plunge-frozen in liquid ethane using the Cryoplunge 3 system (Gatan) operating at 80% humidity, 298K ambient temperature.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Details | In order to account for highly preferred orientations of the specimen, data were acquired using tilts of 40 degrees. |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3838 pixel / Digitization - Frames/image: 1-100 / Average electron dose: 82.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 38167 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.5 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 22500 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |