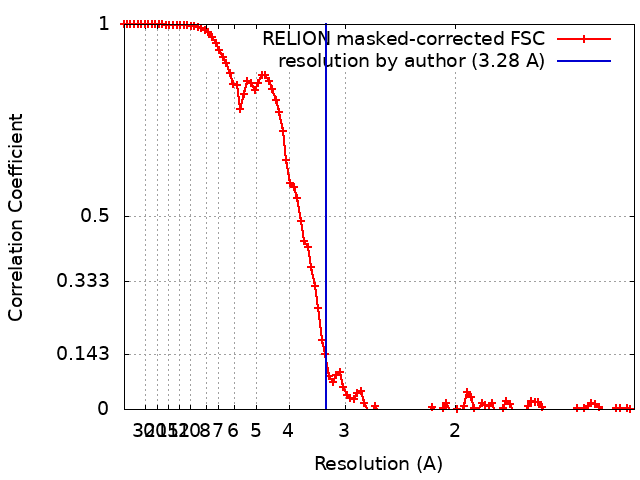
- EMDB-31913: 3.28 A structure of the rabbit muscle aldolase -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: EMDB / ID: EMD-31913
Title
3.28 A structure of the rabbit muscle aldolase
Map data
Sample
Complex: Rabbit muscle aldolase
Protein or peptide: Fructose-bisphosphate aldolase A
Keywords
Rabbit muscle aldolase / STRUCTURAL PROTEIN
Function / homology
Function and homology information
negative regulation of Arp2/3 complex-mediated actin nucleation / fructose-bisphosphate aldolase / fructose-bisphosphate aldolase activity / M band / I band / glycolytic process / protein homotetramerization / positive regulation of cell migration Similarity search - Function
Fructose-bisphosphate aldolase class-I active site / Fructose-bisphosphate aldolase class-I active site. / Fructose-bisphosphate aldolase, class-I / Fructose-bisphosphate aldolase class-I / Aldolase-type TIM barrel Similarity search - Domain/homology
National Natural Science Foundation of China (NSFC)
31830020
China
Chinese Academy of Sciences
XDB37040102
China
National Natural Science Foundation of China (NSFC)
31925026
China
Ministry of Science and Technology (MoST, China)
2015DFG32140
China
Citation
Journal: Nat Commun / Year: 2021 Title: A cryo-electron microscopy support film formed by 2D crystals of hydrophobin HFBI. Authors: Hongcheng Fan / Bo Wang / Yan Zhang / Yun Zhu / Bo Song / Haijin Xu / Yujia Zhai / Mingqiang Qiao / Fei Sun / Abstract: Cryo-electron microscopy (cryo-EM) has become a powerful tool to resolve high-resolution structures of biomacromolecules in solution. However, air-water interface induced preferred orientations, ...Cryo-electron microscopy (cryo-EM) has become a powerful tool to resolve high-resolution structures of biomacromolecules in solution. However, air-water interface induced preferred orientations, dissociation or denaturation of biomacromolecules during cryo-vitrification remains a limiting factor for many specimens. To solve this bottleneck, we developed a cryo-EM support film using 2D crystals of hydrophobin HFBI. The hydrophilic side of the HFBI film adsorbs protein particles via electrostatic interactions and sequesters them from the air-water interface, allowing the formation of sufficiently thin ice for high-quality data collection. The particle orientation distribution can be regulated by adjusting the buffer pH. Using this support, we determined the cryo-EM structures of catalase (2.29 Å) and influenza haemagglutinin trimer (2.56 Å), which exhibited strong preferred orientations using a conventional cryo-vitrification protocol. We further show that the HFBI film is suitable to obtain high-resolution structures of small proteins, including aldolase (150 kDa, 3.28 Å) and haemoglobin (64 kDa, 3.6 Å). Our work suggests that HFBI films may have broad future applications in increasing the success rate and efficiency of cryo-EM.
History
Deposition
Sep 6, 2021
-
Header (metadata) release
Dec 29, 2021
-
Map release
Dec 29, 2021
-
Update
Jun 25, 2025
-
Current status
Jun 25, 2025
Processing site: PDBj / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Authors
Authors China, 5 items
China, 5 items  Citation
Citation Structure visualization
Structure visualization
 Downloads & links
Downloads & links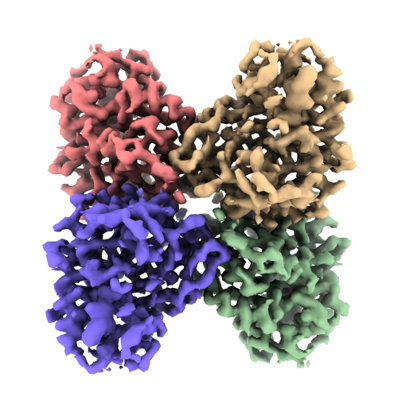 emd_31913.png
emd_31913.png http://ftp.pdbj.org/pub/emdb/structures/EMD-31913
http://ftp.pdbj.org/pub/emdb/structures/EMD-31913




















 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)























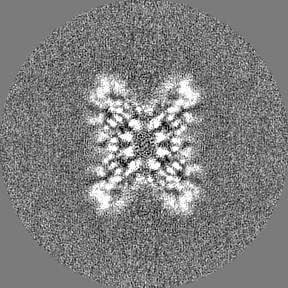
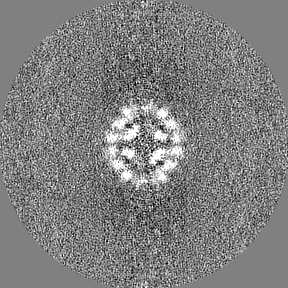






 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN