 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2iw8: STRUCTURE OF HUMAN THR160-PHOSPHO CDK2-CYCLIN A F82H-L83V-H84D MU... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2iw8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF HUMAN THR160-PHOSPHO CDK2-CYCLIN A F82H-L83V-H84D MUTANT WITH AN O6-CYCLOHEXYLMETHYLGUANINE INHIBITOR | ||||||
 Components Components |
| ||||||
 Keywords Keywords | CELL CYCLE / PHOSPHORYLATION / NUCLEOTIDE-BINDING / SERINE/THREONINE-PROTEIN KINASE / CELL CYCLE COMPLEX / KINASE / MITOSIS / TRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationG2 Phase / Phosphorylation of proteins involved in the G2/M transition by Cyclin A:Cdc2 complexes / cyclin A2-CDK1 complex / cyclin A2-CDK2 complex / cell cycle G1/S phase transition / cellular response to luteinizing hormone stimulus / p53-Dependent G1 DNA Damage Response / mitotic cell cycle phase transition / Transcription of E2F targets under negative control by p107 (RBL1) and p130 (RBL2) in complex with HDAC1 / Regulation of APC/C activators between G1/S and early anaphase ...G2 Phase / Phosphorylation of proteins involved in the G2/M transition by Cyclin A:Cdc2 complexes / cyclin A2-CDK1 complex / cyclin A2-CDK2 complex / cell cycle G1/S phase transition / cellular response to luteinizing hormone stimulus / p53-Dependent G1 DNA Damage Response / mitotic cell cycle phase transition / Transcription of E2F targets under negative control by p107 (RBL1) and p130 (RBL2) in complex with HDAC1 / Regulation of APC/C activators between G1/S and early anaphase / cellular response to leptin stimulus / male pronucleus / Telomere Extension By Telomerase / G0 and Early G1 / female pronucleus / response to glucagon / cellular response to cocaine / cellular response to nitric oxide / cyclin-dependent protein serine/threonine kinase regulator activity / cellular response to insulin-like growth factor stimulus / positive regulation of DNA biosynthetic process / TP53 Regulates Transcription of Genes Involved in G1 Cell Cycle Arrest / cochlea development / cellular response to platelet-derived growth factor stimulus / Cyclin A/B1/B2 associated events during G2/M transition / Cyclin A:Cdk2-associated events at S phase entry / cyclin-dependent protein kinase holoenzyme complex / regulation of DNA replication / animal organ regeneration / post-translational protein modification / cellular response to estradiol stimulus / DNA Damage/Telomere Stress Induced Senescence / CDK-mediated phosphorylation and removal of Cdc6 / Cdc20:Phospho-APC/C mediated degradation of Cyclin A / SCF(Skp2)-mediated degradation of p27/p21 / Orc1 removal from chromatin / G1/S transition of mitotic cell cycle / G2/M transition of mitotic cell cycle / positive regulation of fibroblast proliferation / Regulation of TP53 Degradation / Processing of DNA double-strand break ends / Senescence-Associated Secretory Phenotype (SASP) / cellular response to hypoxia / Regulation of TP53 Activity through Phosphorylation / Ras protein signal transduction / Ub-specific processing proteases / protein domain specific binding / cell division / centrosome / DNA-templated transcription / positive regulation of DNA-templated transcription / protein kinase binding / nucleoplasm / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Pratt, D.J. / Bentley, J. / Jewsbury, P. / Boyle, F.T. / Endicott, J.A. / Noble, M.E.M. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2006 Title: Dissecting the Determinants of Cyclin-Dependent Kinase 2 and Cyclin-Dependent Kinase 4 Inhibitor Selectivity. Authors: Pratt, D.J. / Bentley, J. / Jewsbury, P. / Boyle, F.T. / Endicott, J.A. / Noble, M.E.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2iw8.cif.gz | 231.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2iw8.ent.gz | 186.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2iw8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 2iw8_validation.pdf.gz | 1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 2iw8_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 2iw8_validation.xml.gz | 42.8 KB | Display | |
| Data in CIF | 2iw8_validation.cif.gz | 60.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/iw/2iw8ftp://data.pdbj.org/pub/pdb/validation_reports/iw/2iw8 | HTTPS FTP |
-Related structure data
| Related structure data |  2iw6C  2iw9C  1h1sS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






























































































































-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS ensembles :
|
-Components
| #1: Protein | Mass: 34308.652 Da / Num. of mol.: 2 / Mutation: YES Source method: isolated from a genetically manipulated source Details: PHOSPHOTHREONINE 160 / Source: (gene. exp.) HOMO SAPIENS (human) / Description: WILD TYPE GENE WAS A GIFT FROM DR D.O.MORGAN / Plasmid: PGEX 6P-1 / Production host:  #2: Protein | Mass: 29884.605 Da / Num. of mol.: 2 / Fragment: A3, RESIDUES 174-432 Source method: isolated from a genetically manipulated source Details: MONOTHIOGLYCEROL ADDUCT ON RESIDUE 193 / Source: (gene. exp.) HOMO SAPIENS (human)Description: FULL LENGTH CYCLIN A WAS A GIFT FROM DR M.DOREE Plasmid: PET21D / Production host: #3: Chemical | 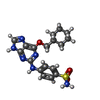  Mass: 402.471 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H22N6O3S Mass: 402.471 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H22N6O3S#4: Chemical |   Mass: 108.159 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2S Mass: 108.159 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 309 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 309 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, PHE 82 TO HIS ENGINEERED RESIDUE IN CHAIN A, LEU 83 TO VAL ...ENGINEERED | Sequence details | GPGS INTRODUCED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.93 Å3/Da / Density % sol: 57.75 % |
|---|---|
| Crystal grow | pH: 7 Details: DROPLET CONTAINED PROTEIN AT A CONCENTRATION OF 10 MG ML-1 IN 40 MM HEPES PH 7.0 CONTAINING 1 MM DTT, 200 MM NACL, 5% V/V DMSO AND 1MM INHIBITOR. THE RESERVOIR SOLUTION CONTAINED 0.8 M KCL ...Details: DROPLET CONTAINED PROTEIN AT A CONCENTRATION OF 10 MG ML-1 IN 40 MM HEPES PH 7.0 CONTAINING 1 MM DTT, 200 MM NACL, 5% V/V DMSO AND 1MM INHIBITOR. THE RESERVOIR SOLUTION CONTAINED 0.8 M KCL AND 1.2 M (NH4)2SO4 IN 40 MM HEPES PH 7.0 AND 5 MM DTT. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 / Beamline: ID14-2 / Wavelength: 0.933 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Nov 21, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→65.9 Å / Num. obs: 64531 / % possible obs: 97.1 % / Observed criterion σ(I): 0 / Redundancy: 2.6 % / Rmerge(I) obs: 0.13 / Net I/σ(I): 3.6 |
| Reflection shell | Resolution: 2.3→2.41 Å / Redundancy: 2.5 % / Rmerge(I) obs: 0.32 / Mean I/σ(I) obs: 2.3 / % possible all: 94.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1H1S Resolution: 2.3→100 Å / Cor.coef. Fo:Fc: 0.928 / Cor.coef. Fo:Fc free: 0.895 / SU B: 14.391 / SU ML: 0.164 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.301 / ESU R Free: 0.231 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 14.38 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→100 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|