 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h00: CDK2 in complex with a disubstituted 4, 6-bis anilino pyrimidine ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h00 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CDK2 in complex with a disubstituted 4, 6-bis anilino pyrimidine CDK4 inhibitor | ||||||
 Components Components | CELL DIVISION PROTEIN KINASE 2 | ||||||
 Keywords Keywords | TRANSFERASE / SERINE/THREONINE-PROTEIN KINASE / MITOSIS | ||||||
| Function / homology |  Function and homology information Function and homology informationcyclin A1-CDK2 complex / cyclin E2-CDK2 complex / cyclin E1-CDK2 complex / cyclin A2-CDK2 complex / G2 Phase / Y chromosome / cyclin-dependent protein kinase activity / regulation of heterochromatin organization / Phosphorylation of proteins involved in G1/S transition by active Cyclin E:Cdk2 complexes / positive regulation of heterochromatin formation ...cyclin A1-CDK2 complex / cyclin E2-CDK2 complex / cyclin E1-CDK2 complex / cyclin A2-CDK2 complex / G2 Phase / Y chromosome / cyclin-dependent protein kinase activity / regulation of heterochromatin organization / Phosphorylation of proteins involved in G1/S transition by active Cyclin E:Cdk2 complexes / positive regulation of heterochromatin formation / p53-Dependent G1 DNA Damage Response / X chromosome / PTK6 Regulates Cell Cycle / regulation of anaphase-promoting complex-dependent catabolic process / Defective binding of RB1 mutants to E2F1,(E2F2, E2F3) / centriole replication / Regulation of APC/C activators between G1/S and early anaphase / telomere maintenance in response to DNA damage / G0 and Early G1 / centrosome duplication / Telomere Extension By Telomerase / Activation of the pre-replicative complex / cyclin-dependent kinase / cyclin-dependent protein serine/threonine kinase activity / TP53 Regulates Transcription of Genes Involved in G1 Cell Cycle Arrest / Regulation of MITF-M-dependent genes involved in cell cycle and proliferation / Cajal body / Activation of ATR in response to replication stress / Cyclin E associated events during G1/S transition / cyclin-dependent protein kinase holoenzyme complex / Cyclin A:Cdk2-associated events at S phase entry / Cyclin A/B1/B2 associated events during G2/M transition / condensed chromosome / mitotic G1 DNA damage checkpoint signaling / cellular response to nitric oxide / negative regulation of protein localization to chromatin / post-translational protein modification / regulation of mitotic cell cycle / cyclin binding / positive regulation of DNA replication / peptidyl-serine phosphorylation / G1/S transition of mitotic cell cycle / meiotic cell cycle / DNA Damage/Telomere Stress Induced Senescence / cellular senescence / G2/M transition of mitotic cell cycle / Meiotic recombination / CDK-mediated phosphorylation and removal of Cdc6 / Transcriptional regulation of granulopoiesis / SCF(Skp2)-mediated degradation of p27/p21 / Orc1 removal from chromatin / Cyclin D associated events in G1 / Regulation of TP53 Degradation / nuclear envelope / Factors involved in megakaryocyte development and platelet production / transcription regulator complex / Processing of DNA double-strand break ends / Senescence-Associated Secretory Phenotype (SASP) / Regulation of TP53 Activity through Phosphorylation / Ras protein signal transduction / ciliary basal body / protein phosphorylation / chromosome, telomeric region / DNA replication / endosome / chromatin remodeling / protein domain specific binding / protein serine kinase activity / cell division / DNA repair / protein serine/threonine kinase activity / positive regulation of cell population proliferation / centrosome / magnesium ion binding / signal transduction / nucleoplasm / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Beattie, J.F. / Breault, G.A. / Ellston, R.P.A. / Green, S. / Jewsbury, P.J. / Midgley, C.J. / Naven, R.T. / Minshull, C.A. / Pauptit, R.A. / Tucker, J.A. / Pease, J.E. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2003 Title: Cyclin-Dependent Kinase 4 Inhibitors as a Treatment for Cancer. Part 1: Identification and Optimisation of Substituted 4,6-Bis Anilino Pyrimidines Authors: Beattie, J.F. / Breault, G.A. / Ellston, R.P.A. / Green, S. / Jewsbury, P.J. / Midgley, C.J. / Naven, R.T. / Minshull, C.A. / Pauptit, R.A. / Tucker, J.A. / Pease, J.E. #1: Journal: J.Med.Chem. / Year: 1996Title: High-Resolution Crystal Structures of Human Cyclin-Dependent Kinase 2 with and without ATP: Bound Waters and Natural Ligand as a Guide for Inhibitor Design Authors: Schulze-Gahmen, U. / De Bondt, H. / Kim, S.-H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h00.cif.gz | 77.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h00.ent.gz | 56.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1h00.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h0/1h00ftp://data.pdbj.org/pub/pdb/validation_reports/h0/1h00 | HTTPS FTP |
|---|
-Related structure data




















































-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34002.527 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:   SPODOPTERA FRUGIPERDA (fall armyworm) / Strain (production host): SF9 / References: UniProt: P24941, EC: 2.7.1.37 SPODOPTERA FRUGIPERDA (fall armyworm) / Strain (production host): SF9 / References: UniProt: P24941, EC: 2.7.1.37 |
|---|---|
| #2: Chemical | ChemComp-FAP / (  Mass: 415.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H23F2N5O2 Mass: 415.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H23F2N5O2 |
| #3: Chemical | ChemComp-FCP / (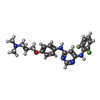  Mass: 415.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H23F2N5O2 Mass: 415.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H23F2N5O2 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 209 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 209 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 39 % |
|---|---|
| Crystal grow | pH: 7 Details: PROTEIN AT 10MG/ML WELL BUFFER CONTAINING 17.5% PEG3350, 200MM HEPES, PH7.0, 100MM AMMONIUM ACETATE, pH 7.00 |
| Crystal grow | *PLUS Method: unknown / Details: Lawrie, A.M., (1997) Nat.Struct.Biol., 4, 796. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX14.2 / Wavelength: 0.978 / Beamline: PX14.2 / Wavelength: 0.978 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Feb 8, 2002 |
| Radiation | Monochromator: SI(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.978 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→32.3 Å / Num. obs: 106279 / % possible obs: 93.6 % / Redundancy: 3 % / Biso Wilson estimate: 22.3 Å2 / Rmerge(I) obs: 0.036 / Net I/σ(I): 20.9 |
| Reflection shell | Resolution: 1.6→1.69 Å / Redundancy: 1.6 % / Rmerge(I) obs: 0.372 / Mean I/σ(I) obs: 2 / % possible all: 69.7 |
| Reflection | *PLUS Highest resolution: 1.6 Å / Num. obs: 35229 / Num. measured all: 106279 / Rmerge(I) obs: 0.036 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.6→32.3 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 1274839.6 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: RESIDUES 13 - 14, 36 - 43, AND 152 - 161 ARE NOT VISIBLE IN THE ELECTRON DENSITY MAP.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 53.782 Å2 / ksol: 0.370039 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→32.3 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.7 Å / Rfactor Rfree error: 0.024 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.6 Å / % reflection Rfree: 5 % / Rfactor Rfree: 0.25 / Rfactor Rwork: 0.21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
