 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2cgu: Identification of chemically diverse Chk1 inhibitors by receptor-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2cgu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Identification of chemically diverse Chk1 inhibitors by receptor- based virtual screening | ||||||
 Components Components | SERINE/THREONINE-PROTEIN KINASE CHK1 | ||||||
 Keywords Keywords | TRANSFERASE / DOCKING / DRUG DESIGN / ONCOLOGY / VIRTUAL SCREENING / ATP- BINDING / CELL CYCLE / DNA DAMAGE / DNA REPAIR / KINASE / NUCLEAR PROTEIN / NUCLEOTIDE-BINDING / PHOSPHORYLATION / POLYMORPHISM / SERINE/THREONINE-PROTEIN KINASE / UBL CONJUGATION | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of mitotic nuclear division / apoptotic process involved in development / negative regulation of G0 to G1 transition / histone H3T11 kinase activity / regulation of mitotic centrosome separation / mitotic G2/M transition checkpoint / peptidyl-threonine phosphorylation / inner cell mass cell proliferation / regulation of double-strand break repair via homologous recombination / nucleus organization ...negative regulation of mitotic nuclear division / apoptotic process involved in development / negative regulation of G0 to G1 transition / histone H3T11 kinase activity / regulation of mitotic centrosome separation / mitotic G2/M transition checkpoint / peptidyl-threonine phosphorylation / inner cell mass cell proliferation / regulation of double-strand break repair via homologous recombination / nucleus organization / negative regulation of gene expression, epigenetic / mitotic G2 DNA damage checkpoint signaling / Transcriptional Regulation by E2F6 / replicative senescence / Presynaptic phase of homologous DNA pairing and strand exchange / Activation of ATR in response to replication stress / Chk1/Chk2(Cds1) mediated inactivation of Cyclin B:Cdk1 complex / signal transduction in response to DNA damage / positive regulation of cell cycle / DNA damage checkpoint signaling / replication fork / regulation of signal transduction by p53 class mediator / condensed nuclear chromosome / TP53 Regulates Transcription of DNA Repair Genes / cellular response to mechanical stimulus / Signaling by SCF-KIT / G2/M DNA damage checkpoint / Ubiquitin-Mediated Degradation of Phosphorylated Cdc25A / G2/M transition of mitotic cell cycle / regulation of cell population proliferation / Processing of DNA double-strand break ends / Regulation of TP53 Activity through Phosphorylation / protein phosphorylation / protein kinase activity / DNA replication / non-specific serine/threonine protein kinase / chromatin remodeling / protein domain specific binding / protein serine kinase activity / DNA repair / protein serine/threonine kinase activity / apoptotic process / DNA damage response / centrosome / chromatin / protein-containing complex / : / nucleoplasm / ATP binding / nucleus / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Foloppe, N. / Fisher, L.M. / Howes, R. / Potter, A. / Robertson, A.G.S. / Surgenor, A.E. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem. / Year: 2006 Title: Identification of Chemically Diverse Chk1 Inhibitors by Receptor-Based Virtual Screening. Authors: Foloppe, N. / Fisher, L.M. / Howes, R. / Potter, A. / Robertson, A.G.S. / Surgenor, A.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2cgu.cif.gz | 72.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2cgu.ent.gz | 52.9 KB | Display | PDB format |
| PDBx/mmJSON format | 2cgu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cg/2cguftp://data.pdbj.org/pub/pdb/validation_reports/cg/2cgu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2cgvC  2cgwC  2cgxC  1ia8S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




























-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 34148.148 Da / Num. of mol.: 1 / Fragment: N-TERMINAL KINASE DOMAIN, RESIDUES 1-289 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PFASTBAC1/CHK1 1-289 C8H / Cell line (production host): SF9 / Production host:   SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: O14757, EC: 2.7.1.37 SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: O14757, EC: 2.7.1.37 |
|---|---|
| #2: Chemical | ChemComp-3A3 / 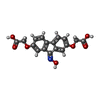  Mass: 343.288 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H13NO7 Mass: 343.288 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C17H13NO7 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 156 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 156 / Source method: isolated from a natural source / Formula: H2O |
| Compound details | REQUIRED FOR CHECKPOINT MEDIATED CELL CYCLE ARREST IN RESPONSE TO DNA DAMAGE OR THE PRESENCE OF ...REQUIRED FOR CHECKPOINT |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 56.9 % |
|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.50 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.9792 / Beamline: 19-ID / Wavelength: 0.9792 |
| Detector | Type: ADSC CCD / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9792 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→30 Å / Num. obs: 11961 / % possible obs: 70.5 % / Observed criterion σ(I): 2 / Redundancy: 4.1 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 10.6 |
| Reflection shell | Resolution: 2.5→2.57 Å / Redundancy: 0.9 % / Rmerge(I) obs: 0.23 / Mean I/σ(I) obs: 2.3 / % possible all: 22.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1IA8 Resolution: 2.5→30 Å / Cor.coef. Fo:Fc: 0.944 / Cor.coef. Fo:Fc free: 0.872 / SU B: 12.875 / SU ML: 0.267 / Cross valid method: THROUGHOUT / ESU R Free: 0.416 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 42.26 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|