 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6qtk: 2.31A structure of gepotidacin with S.aureus DNA gyrase and doubl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6qtk | ||||||
|---|---|---|---|---|---|---|---|
| Title | 2.31A structure of gepotidacin with S.aureus DNA gyrase and doubly nicked DNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | ISOMERASE / Inhibitor / DNA / Complex | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase (ATP-hydrolysing) / DNA topological change / DNA-templated DNA replication / chromosome / response to antibiotic / DNA binding / ATP binding / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria)synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.31 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.31 Å | ||||||
 Authors Authors | Bax, B.D. | ||||||
 Citation Citation | Journal: Acs Infect Dis. / Year: 2019 Title: Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase. Authors: Gibson, E.G. / Bax, B. / Chan, P.F. / Osheroff, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6qtk.cif.gz | 332.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6qtk.ent.gz | 260.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6qtk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qt/6qtkftp://data.pdbj.org/pub/pdb/validation_reports/qt/6qtk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6qtpC  2xcsS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: _ / Refine code: _
NCS ensembles :
|
-Components
-DNA gyrase subunit ... , 2 types, 4 molecules BDAC
| #1: Protein | Mass: 22605.689 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (strain N315) (bacteria)Strain: N315 / Gene: gyrB, SA0005 / Production host: #2: Protein | Mass: 55521.312 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Gene: gyrA, SA0006 / Production host: |
|---|
-DNA chain , 1 types, 2 molecules EF
| #3: DNA chain | Mass: 6134.967 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host: synthetic construct (others) |
|---|
-Non-polymers , 5 types, 835 molecules 


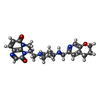

| #4: Chemical |  Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn#5: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C3H8O3#6: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#7: Chemical | ChemComp-JHN / ( |  Mass: 448.518 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H28N6O3 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication, antibiotic, inhibitor*YM Mass: 448.518 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C24H28N6O3 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication, antibiotic, inhibitor*YM#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 823 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.01 Å3/Da / Density % sol: 59.14 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: microbatch / Details: 11% PEG 5000MME, BisTris pH 6.2 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 0.8726 Å / Beamline: ID23-2 / Wavelength: 0.8726 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Sep 28, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.8726 Å / Relative weight: 1 |
| Reflection | Resolution: 2.31→40 Å / Num. obs: 85905 / % possible obs: 99.7 % / Redundancy: 5.5 % / Net I/σ(I): 19.2 |
| Reflection shell | Resolution: 2.31→2.35 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: 2XCS Resolution: 2.31→39.99 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.939 / SU B: 5.843 / SU ML: 0.137 / Cross valid method: THROUGHOUT / ESU R: 0.244 / ESU R Free: 0.188
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.9 Å / Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.747 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.31→39.99 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|