 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2xcs: The 2.1A crystal structure of S. aureus Gyrase complex with GSK29... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2xcs | ||||||
|---|---|---|---|---|---|---|---|
| Title | The 2.1A crystal structure of S. aureus Gyrase complex with GSK299423 and DNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | ISOMERASE / TYPE IIA TOPOISOMERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase (ATP-hydrolysing) / DNA topological change / DNA-templated DNA replication / chromosome / response to antibiotic / DNA binding / ATP binding / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   STAPHYLOCOCCUS AUREUS (bacteria) STAPHYLOCOCCUS AUREUS (bacteria)SYNTHETIC CONSTRUCT (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Bax, B.D. / Chan, P.F. / Eggleston, D.S. / Fosberry, A. / Gentry, D.R. / Gorrec, F. / Giordano, I. / Hann, M.M. / Hennessy, A. / Hibbs, M. ...Bax, B.D. / Chan, P.F. / Eggleston, D.S. / Fosberry, A. / Gentry, D.R. / Gorrec, F. / Giordano, I. / Hann, M.M. / Hennessy, A. / Hibbs, M. / Huang, J. / Jones, E. / Jones, J. / Brown, K.K. / Lewis, C.J. / May, E.W. / Singh, O. / Spitzfaden, C. / Shen, C. / Shillings, A. / Theobald, A.F. / Wohlkonig, A. / Pearson, N.D. / Gwynn, M.N. | ||||||
 Citation Citation | Journal: Nature / Year: 2010 Title: Type Iia Topoisomerase Inhibition by a New Class of Antibacterial Agents. Authors: Bax, B.D. / Chan, P.F. / Eggleston, D.S. / Fosberry, A. / Gentry, D.R. / Gorrec, F. / Giordano, I. / Hann, M.M. / Hennessy, A. / Hibbs, M. / Huang, J. / Jones, E. / Jones, J. / Brown, K.K. / ...Authors: Bax, B.D. / Chan, P.F. / Eggleston, D.S. / Fosberry, A. / Gentry, D.R. / Gorrec, F. / Giordano, I. / Hann, M.M. / Hennessy, A. / Hibbs, M. / Huang, J. / Jones, E. / Jones, J. / Brown, K.K. / Lewis, C.J. / May, E.W. / Saunders, M.R. / Singh, O. / Spitzfaden, C. / Shen, C. / Shillings, A. / Theobald, A.F. / Wohlkonig, A. / Pearson, N.D. / Gwynn, M.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2xcs.cif.gz | 325.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2xcs.ent.gz | 257.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2xcs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/xc/2xcsftp://data.pdbj.org/pub/pdb/validation_reports/xc/2xcs | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2xcoC  2xcqSC  2xcrC  2xctC C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 78109.000 Da / Num. of mol.: 2 Fragment: GYRB- C-TERMINAL 27KDA DOMAIN, RESIDUES 410-543 AND 580-644, GYRA- N-TERMINAL 56KDA DOMAIN, RESIDUES 2-491 Mutation: YES Source method: isolated from a genetically manipulated source Details: C-TERMINUS GYRB (644) FUSED TO N-TERMINUS GYRA (1002). GREEK KEY DOMAIN (544-579) DELETED AND REPLACED WITH TWO RESIDUES, TG Source: (gene. exp.) STAPHYLOCOCCUS AUREUS (bacteria) / Strain: N315 / Production host: #2: DNA chain | Mass: 6099.983 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) SYNTHETIC CONSTRUCT (others) #3: Chemical | ChemComp-MN /   Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn Mass: 54.938 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mn#4: Chemical | ChemComp-RXV / | 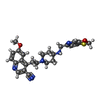  Mass: 461.579 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H27N5O2S / Comment: antibiotic*YM Mass: 461.579 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C25H27N5O2S / Comment: antibiotic*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 826 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 826 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED | Nonpolymer details | MANGANESE ION (MN): MN2+ ION | Sequence details | GREEK KEY DOMAIN (544-579) DELETED AND REPLACED WITH TWO RESIDUES, TG CATALYTIC TYROSINE (1123) ...GREEK KEY DOMAIN (544-579) DELETED AND REPLACED WITH TWO RESIDUES, TG CATALYTIC TYROSINE (1123) MUTATED TO PHENYLALAN | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.92 Å3/Da / Density % sol: 57.48 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.5 / Details: 18% PEG 5000MME, 0.1 M BISTRIS PH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 0.9791 / Beamline: ID23-1 / Wavelength: 0.9791 |
| Detector | Type: MARRESEARCH / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9791 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→30 Å / Num. obs: 113168 / % possible obs: 98.6 % / Observed criterion σ(I): 0 / Redundancy: 5 % / Biso Wilson estimate: 28.25 Å2 / Rmerge(I) obs: 0.12 / Net I/σ(I): 11.4 |
| Reflection shell | Resolution: 2.1→2.21 Å / Redundancy: 4.3 % / Rmerge(I) obs: 0.45 / Mean I/σ(I) obs: 2.6 / % possible all: 98.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2XCQ Resolution: 2.1→20 Å / Cor.coef. Fo:Fc: 0.949 / Cor.coef. Fo:Fc free: 0.938 / SU B: 4.114 / SU ML: 0.112 / Cross valid method: THROUGHOUT / ESU R: 0.175 / ESU R Free: 0.152 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY. COMPOUND SITS ON TWOFOLD AXIS OF THE DIMER. TO SEE BINDING MODE OF SINGLE COMPOUND DELETE B CONFORMERS OF ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY. COMPOUND SITS ON TWOFOLD AXIS OF THE DIMER. TO SEE BINDING MODE OF SINGLE COMPOUND DELETE B CONFORMERS OF COMPOUND AND SIDE-CHAINS B83, B121, D83, D121.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 38.417 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|