 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4z3c | ||||||
|---|---|---|---|---|---|---|---|
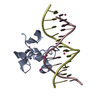
| Title | Zinc finger region of human TET3 in complex with CpG DNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA Binding protein/DNA / zinc finger / dna-binding / Structural Genomics / Structural Genomics Consortium / SGC / DNA Binding protein-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationepigenetic programing of male pronucleus / methylcytosine dioxygenase / DNA 5-methylcytosine dioxygenase activity / TET1,2,3 and TDG demethylate DNA / chromosomal 5-methylcytosine DNA demethylation, oxidation pathway / positive regulation of gene expression via chromosomal CpG island demethylation / protein O-linked glycosylation / male pronucleus / methyl-CpG binding / female pronucleus ...epigenetic programing of male pronucleus / methylcytosine dioxygenase / DNA 5-methylcytosine dioxygenase activity / TET1,2,3 and TDG demethylate DNA / chromosomal 5-methylcytosine DNA demethylation, oxidation pathway / positive regulation of gene expression via chromosomal CpG island demethylation / protein O-linked glycosylation / male pronucleus / methyl-CpG binding / female pronucleus / Chromatin modifications during the maternal to zygotic transition (MZT) / chromosome / RNA polymerase II cis-regulatory region sequence-specific DNA binding / positive regulation of transcription by RNA polymerase II / zinc ion binding / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.57 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.57 Å | ||||||
 Authors Authors | Liu, K. / Xu, C. / Tempel, W. / Dong, A. / Arrowsmith, C.H. / Bountra, C. / Edwards, A.M. / Min, J. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Structure / Year: 2017 Title: DNA Sequence Recognition of Human CXXC Domains and Their Structural Determinants. Authors: Xu, C. / Liu, K. / Lei, M. / Yang, A. / Li, Y. / Hughes, T.R. / Min, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4z3c.cif.gz | 64.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4z3c.ent.gz | 43 KB | Display | PDB format |
| PDBx/mmJSON format | 4z3c.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 4z3c_validation.pdf.gz | 433.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 4z3c_full_validation.pdf.gz | 433.5 KB | Display | |
| Data in XML | 4z3c_validation.xml.gz | 6 KB | Display | |
| Data in CIF | 4z3c_validation.cif.gz | 7.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/z3/4z3cftp://data.pdbj.org/pub/pdb/validation_reports/z3/4z3c | HTTPS FTP |
-Related structure data
| Related structure data |  4nw3C  4o64C  4pziC  5vc9C  5w9qC  5w9sC  6asbC  6asdC  4hp3S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 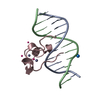
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 3663.392 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: synthetic / Source: (synth.) synthetic construct (others) #2: Protein | | Mass: 5897.102 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: TET3 / Plasmid: pET28-MHL / Production host:  #3: Chemical | ChemComp-UNX /   Num. of mol.: 4 / Source method: obtained synthetically Num. of mol.: 4 / Source method: obtained synthetically#4: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 85 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48.3 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion / pH: 7.5 / Details: 30% PEG-1500, 0.2 M sodium chloride, 0.1 M HEPES |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97918 Å / Beamline: 19-ID / Wavelength: 0.97918 Å | |||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jun 20, 2013 | |||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97918 Å / Relative weight: 1 | |||||||||||||||||||||||||||
| Reflection | Resolution: 1.57→39.11 Å / Num. obs: 16318 / % possible obs: 99.7 % / Redundancy: 3.6 % / CC1/2: 0.998 / Rmerge(I) obs: 0.07 / Rpim(I) all: 0.042 / Net I/σ(I): 10.4 / Num. measured all: 59473 | |||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4HP3 Resolution: 1.57→39.11 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.932 / WRfactor Rfree: 0.2466 / WRfactor Rwork: 0.2132 / FOM work R set: 0.7594 / SU B: 6.238 / SU ML: 0.095 / SU R Cruickshank DPI: 0.097 / SU Rfree: 0.0994 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.097 / ESU R Free: 0.099 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: the structure was by molecular replacement using diffraction data collected on an isomorphous sanple. coot was used for interactive model building. Model geometry was assessed with phenix.molprobity.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 54.84 Å2 / Biso mean: 28.058 Å2 / Biso min: 16.44 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.57→39.11 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.57→1.611 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -3.1291 Å / Origin y: 9.615 Å / Origin z: -11.6993 Å
|
