 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5w9s | ||||||
|---|---|---|---|---|---|---|---|
| Title | Zinc finger of human CXXC5 in complex with CpG DNA | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA BINDING PROTEIN/DNA / Structural Genomics Consortium / SGC / DNA BINDING PROTEIN-DNA complex | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of generation of precursor metabolites and energy / methyl-CpG binding / Estrogen-dependent gene expression / DNA-binding transcription factor binding / sequence-specific DNA binding / positive regulation of canonical NF-kappaB signal transduction / negative regulation of transcription by RNA polymerase II / nucleoplasm / zinc ion binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Liu, K. / Xu, C. / Tempel, W. / Walker, J.R. / Bountra, C. / Arrowsmith, C.H. / Edwards, A.M. / Min, J. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Structure / Year: 2018 Title: DNA Sequence Recognition of Human CXXC Domains and Their Structural Determinants. Authors: Xu, C. / Liu, K. / Lei, M. / Yang, A. / Li, Y. / Hughes, T.R. / Min, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5w9s.cif.gz | 62.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5w9s.ent.gz | 41.9 KB | Display | PDB format |
| PDBx/mmJSON format | 5w9s.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/w9/5w9sftp://data.pdbj.org/pub/pdb/validation_reports/w9/5w9s | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4nw3C  4o64C  4pziC  4z3cC  5vc9C  5w9qC  6asbC  6asdC  4hp3S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 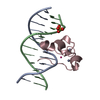
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-DNA chain / Protein , 2 types, 3 molecules ABC
| #1: DNA chain | Mass: 3663.392 Da / Num. of mol.: 2 / Source method: obtained synthetically / Details: synthetic / Source: (synth.) synthetic construct (others) #2: Protein | | Mass: 8335.792 Da / Num. of mol.: 1 / Fragment: Zinc finger domain (UNP residues 254-306) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CXXC5, HSPC195, TCCCIA00297 / Plasmid: pET28-MHL / Production host:  |
|---|
-Non-polymers , 4 types, 16 molecules 



| #3: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-SO4 / |  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-UNX / |  Num. of mol.: 1 / Source method: obtained synthetically Num. of mol.: 1 / Source method: obtained synthetically#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 12 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 48.91 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion / pH: 6.5 / Details: 30% PEG-5000-MME, 0.2M ammonium sulfate, 0.1M MES |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E SUPERBRIGHT / Wavelength: 1.5418 Å | ||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Aug 22, 2012 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.1→34.74 Å / Num. obs: 8535 / % possible obs: 100 % / Redundancy: 11.3 % / CC1/2: 1 / Rmerge(I) obs: 0.04 / Rpim(I) all: 0.012 / Rrim(I) all: 0.042 / Net I/σ(I): 34.9 / Num. measured all: 96584 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: _
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 4hp3 Resolution: 2.1→34.74 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.951 / SU B: 11.434 / SU ML: 0.156 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.2 / ESU R Free: 0.176 Details: The amino acid sequence of the search model was adjusted with Chainsaw after molecular replacement. Coot was used for interactive model re-building. molprobity and the parvati server were ...Details: The amino acid sequence of the search model was adjusted with Chainsaw after molecular replacement. Coot was used for interactive model re-building. molprobity and the parvati server were used for the analysis of model geometry and anisotropic ADPs, respectively.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 83.14 Å2 / Biso mean: 49.286 Å2 / Biso min: 23.47 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.1→34.74 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.101→2.156 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
