 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2cjz: crystal structure of the c472s mutant of human protein tyrosine p... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2cjz | ||||||
|---|---|---|---|---|---|---|---|
| Title | crystal structure of the c472s mutant of human protein tyrosine phosphatase ptpn5 (step, striatum enriched phosphatase) in complex with phosphotyrosine | ||||||
 Components Components | HUMAN PROTEIN TYROSINE PHOSPHATASE PTPN5 | ||||||
 Keywords Keywords | HYDROLASE / PROTEIN PHOSPHATASE / STEP / PTPN5 / PHOSPHATASE | ||||||
| Function / homology |  Function and homology information Function and homology informationInterleukin-37 signaling / protein dephosphorylation / phosphotyrosine residue binding / protein-tyrosine-phosphatase / protein tyrosine phosphatase activity / cell junction / protein kinase binding / endoplasmic reticulum membrane / signal transduction / nucleoplasm ...Interleukin-37 signaling / protein dephosphorylation / phosphotyrosine residue binding / protein-tyrosine-phosphatase / protein tyrosine phosphatase activity / cell junction / protein kinase binding / endoplasmic reticulum membrane / signal transduction / nucleoplasm / membrane / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Debreczeni, J.E. / Barr, A.J. / Eswaran, J. / Smee, C. / Burgess, N. / Gileadi, O. / Savitsky, P. / Sundstrom, M. / Arrowsmith, C. / Edwards, A. ...Debreczeni, J.E. / Barr, A.J. / Eswaran, J. / Smee, C. / Burgess, N. / Gileadi, O. / Savitsky, P. / Sundstrom, M. / Arrowsmith, C. / Edwards, A. / Weigelt, J. / Knapp, S. / von Delft, F. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2009 Title: Large-Scale Structural Analysis of the Classical Human Protein Tyrosine Phosphatome. Authors: Barr, A.J. / Ugochukwu, E. / Lee, W.H. / King, O.N.F. / Filippakopoulos, P. / Alfano, I. / Savitsky, P. / Burgess-Brown, N.A. / Muller, S. / Knapp, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2cjz.cif.gz | 76.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2cjz.ent.gz | 54.8 KB | Display | PDB format |
| PDBx/mmJSON format | 2cjz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cj/2cjzftp://data.pdbj.org/pub/pdb/validation_reports/cj/2cjz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2ahsC  2b49C  2cfvC  2gjtC  2h4vC  2i75C  2jjdC  2nlkC  2nz6C  2oc3C  2ooqC  2p6xC  2pa5C  2qepC  3b7oC  2bijS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 35188.777 Da / Num. of mol.: 1 / Fragment: PHOSPHATASE DOMAIN, RESIDUES 258-539 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PLIC SGC / Production host:  | ||
|---|---|---|---|
| #2: Chemical | ChemComp-PTR / 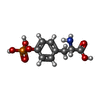  Type: L-peptide linking / Mass: 261.168 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H12NO6P Type: L-peptide linking / Mass: 261.168 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H12NO6P | ||
| #3: Chemical | ChemComp-EDO /   Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 | ||
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 212 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 212 / Source method: isolated from a natural source / Formula: H2O | ||
| Compound details | ENGINEERED| Sequence details | RESIDUES ASP 289, LEU 298, VAL 299 AND THR 517 ARE GIVEN AS VARIANTS IN THE UNIPROT ENTRY P54829 ...RESIDUES ASP 289, LEU 298, VAL 299 AND THR 517 ARE GIVEN AS VARIANTS IN THE UNIPROT ENTRY P54829 AND REFERNCED IN PUBMED ID: 14702039. RESIDUES -23 TO -1 FORM PART OF A N-TERMINAL HIS-TAG USED FOR EXPRESSION | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48 % |
|---|---|
| Crystal grow | Method: vapor diffusion, sitting drop Details: 150 NL SITTING DROPS, 0.1 M CACL2, 0.1 M TRIS PH 8, 20% PEG 6K, 10% ETHYLENE GLYCOL |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 0.95 / Beamline: X10SA / Wavelength: 0.95 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Mar 24, 2006 / Details: MIRRORS |
| Radiation | Monochromator: MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→50.39 Å / Num. obs: 37692 / % possible obs: 98.5 % / Observed criterion σ(I): 3 / Redundancy: 3.4 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 11.92 |
| Reflection shell | Resolution: 1.7→1.8 Å / Redundancy: 2.31 % / Rmerge(I) obs: 0.27 / Mean I/σ(I) obs: 3.07 / % possible all: 91.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2BIJ Resolution: 1.7→101.02 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.939 / SU B: 3.529 / SU ML: 0.06 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.093 / ESU R Free: 0.093 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.38 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→101.02 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|