 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lfo | ||||||
|---|---|---|---|---|---|---|---|
| Title | LIVER FATTY ACID BINDING PROTEIN-OLEATE COMPLEX | ||||||
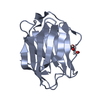
 Components Components | LIVER FATTY ACID BINDING PROTEIN | ||||||
 Keywords Keywords | INTRACELLULAR LIPID TRANSPORT PROTEIN / FATTY ACID BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationacylglycerol metabolic process / digestive system process / monoacylglycerol metabolic process / acylglycerol biosynthetic process / fatty acid primary amide metabolic process / fatty acid derivative metabolic process / endocannabinoid signaling pathway / diterpenoid metabolic process / hepatic stellate cell activation / Regulation of lipid metabolism by PPARalpha ...acylglycerol metabolic process / digestive system process / monoacylglycerol metabolic process / acylglycerol biosynthetic process / fatty acid primary amide metabolic process / fatty acid derivative metabolic process / endocannabinoid signaling pathway / diterpenoid metabolic process / hepatic stellate cell activation / Regulation of lipid metabolism by PPARalpha / chylomicron assembly / long-chain fatty acyl-CoA binding / Triglyceride catabolism / : / lysophospholipid:sodium symporter activity / : / long-chain fatty acid import into cell / connective tissue replacement / Heme degradation / reduction of food intake in response to dietary excess / Cytoprotection by HMOX1 / cannabinoid signaling pathway / intracellular signaling cassette / long-chain fatty acid metabolic process / ketone metabolic process / unsaturated fatty acid biosynthetic process / sterol binding / oleic acid binding / lipid droplet formation / response to tetrachloromethane / glycerol metabolic process / bile acid secretion / adult feeding behavior / D-glucose transmembrane transport / arachidonate metabolic process / aldehyde metabolic process / lipoprotein transport / apical cortex / triglyceride biosynthetic process / positive regulation of fatty acid beta-oxidation / smooth muscle tissue development / very-low-density lipoprotein particle clearance / lipoprotein metabolic process / response to fatty acid / vesicle budding from membrane / L-glutamine metabolic process / bile acid binding / response to cholesterol / bile acid metabolic process / response to vitamin B3 / retinol metabolic process / cellular detoxification / digestive tract development / intestinal absorption / cholesterol efflux / heterocyclic compound binding / triglyceride metabolic process / long-chain fatty acid transmembrane transporter activity / triglyceride homeostasis / cholesterol binding / response to starvation / fatty acid beta-oxidation / response to food / antioxidant activity / response to lipid / fatty acid oxidation / peroxisomal matrix / long-chain fatty acid transport / fatty acid transport / xenobiotic transport / cholesterol metabolic process / energy homeostasis / phospholipid metabolic process / response to nutrient / establishment of localization in cell / fatty acid binding / cholesterol homeostasis / glycolytic process / lipid metabolic process / glutathione metabolic process / liver development / fatty acid metabolic process / phospholipid binding / multicellular organism growth / glucose metabolic process / fatty acid biosynthetic process / cellular response to hydrogen peroxide / response to oxidative stress / cellular response to hypoxia / gene expression / cell population proliferation / response to xenobiotic stimulus / inflammatory response / chromatin binding / lipid binding / protein-containing complex / nucleoplasm / membrane / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Thompson, J. / Winter, N. / Terwey, D. / Bratt, J. / Banaszak, L. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1997 Title: The crystal structure of the liver fatty acid-binding protein. A complex with two bound oleates. Authors: Thompson, J. / Winter, N. / Terwey, D. / Bratt, J. / Banaszak, L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lfo.cif.gz | 41 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lfo.ent.gz | 27.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1lfo.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lf/1lfoftp://data.pdbj.org/pub/pdb/validation_reports/lf/1lfo | HTTPS FTP |
|---|
-Related structure data
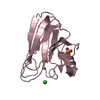
| Related structure data |  1adlS  1cbrS  1crbS  1hmrS  1ifcS  1opbS S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 14333.608 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: AMINO-TERMINAL INITIATOR METHIONINE AND MODIFIED CYSTEINE 69 PRESENT Source: (gene. exp.)  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | 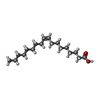  Mass: 282.461 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H34O2 Mass: 282.461 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H34O2#3: Chemical | ChemComp-BEO / |   Mass: 86.089 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O2 Mass: 86.089 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O2#4: Chemical | ChemComp-UNX / |   Num. of mol.: 1 / Source method: obtained synthetically Num. of mol.: 1 / Source method: obtained synthetically#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 61 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 55 % Description: DATA IN THE 2.3 - 2.1 ANGSTROM RANGE WAS NOT USED DUE TO ITS POOR QUALITY. | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 5.6 Details: HANGING DROP VAPOR DIFFUSION EXPERIMENT 1 ML WELL: 3 M AMMONIUM SULFATE, 200 MM LISO4, 100 MM CITRATE, AT A PH OF 5.6 STOCK: 13 MG/ML LFABP-OLEATE COMPLEX 10 MICROLITER DROP: 1:1 MIXTURE OF ...Details: HANGING DROP VAPOR DIFFUSION EXPERIMENT 1 ML WELL: 3 M AMMONIUM SULFATE, 200 MM LISO4, 100 MM CITRATE, AT A PH OF 5.6 STOCK: 13 MG/ML LFABP-OLEATE COMPLEX 10 MICROLITER DROP: 1:1 MIXTURE OF STOCK AND WELL, vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 287 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Aug 26, 1991 |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→38.8 Å / Num. obs: 8640 / % possible obs: 79.3 % / Observed criterion σ(I): 0 / Redundancy: 5.3 % / Biso Wilson estimate: 36.8 Å2 / Rsym value: 0.1 / Net I/σ(I): 10.7 |
| Reflection shell | Resolution: 2.1→2.3 Å / Mean I/σ(I) obs: 0.89 / Rsym value: 0.535 / % possible all: 33.2 |
| Reflection | *PLUS Rmerge(I) obs: 0.1 |
| Reflection shell | *PLUS % possible obs: 33.2 % / Num. unique obs: 940 / Rmerge(I) obs: 0.535 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: SUPERIMPOSED POLYALANINE COMPOSITE STRUCTURE OF 1ADL, 1CBR, 1CRB, 1OPB, 1IFC, 1HMR Resolution: 2.3→8 Å / Rfactor Rfree error: 0.013 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 1
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 40.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.33 Å / Rfactor Rfree error: 0.131 / Total num. of bins used: 30
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
