Entry Database : PDB / ID : 3ar5Title Calcium pump crystal structure with bound TNP-AMP and TG Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 Keywords / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Oryctolagus cuniculus (rabbit)Method / / / Resolution : 2.2 Å Authors Toyoshima, C. / Yonekura, S. / Tsueda, J. / Iwasawa, S. Journal : Proc.Natl.Acad.Sci.USA / Year : 2011Title : Trinitrophenyl derivatives bind differently from parent adenine nucleotides to Ca2+-ATPase in the absence of Ca2+Authors : Toyoshima, C. / Yonekura, S. / Tsueda, J. / Iwasawa, S. History Deposition Nov 24, 2010 Deposition site / Processing site Revision 1.0 Feb 2, 2011 Provider / Type Revision 1.1 Jul 13, 2011 Group Revision 1.2 Jul 31, 2013 Group Revision 1.3 Apr 3, 2024 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Refinement description / Structure summary Category chem_comp / chem_comp_atom ... chem_comp / chem_comp_atom / chem_comp_bond / database_2 / entity / pdbx_entity_nonpoly / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_conn_type / struct_ref_seq_dif / struct_site Item _chem_comp.name / _database_2.pdbx_DOI ... _chem_comp.name / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _entity.pdbx_description / _pdbx_entity_nonpoly.name / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.conn_type_id / _struct_conn.id / _struct_conn.pdbx_dist_value / _struct_conn.pdbx_leaving_atom_flag / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn_type.id / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.4 Oct 23, 2024 Group / Category / pdbx_modification_feature / Item
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






























 PDBj
PDBj
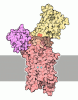
 Assembly
Assembly

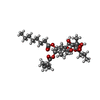
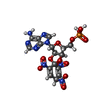


 Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na
Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 650.754 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H50O12
Mass: 650.754 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H50O12 Mass: 558.310 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H15N8O13P
Mass: 558.310 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H15N8O13P Mass: 734.039 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C40H80NO8P / Comment: phospholipid*YM
Mass: 734.039 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C40H80NO8P / Comment: phospholipid*YM Sample preparation
Sample preparation / Beamline: BL41XU / Wavelength: 0.9 Å
/ Beamline: BL41XU / Wavelength: 0.9 Å Processing
Processing