 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5d58: In meso in situ serial X-ray crystallography structure of the Pep... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5d58 | ||||||
|---|---|---|---|---|---|---|---|
| Title | In meso in situ serial X-ray crystallography structure of the PepTSt-Ala-Phe complex at 100 K | ||||||
 Components Components | Di-or tripeptide:H+ symporter | ||||||
 Keywords Keywords | TRANSPORT PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationtripeptide transmembrane transport / tripeptide transmembrane transporter activity / peptide:proton symporter activity / dipeptide transmembrane transporter activity / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Streptococcus thermophilus (bacteria) Streptococcus thermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Huang, C.-Y. / Olieric, V. / Diederichs, K. / Wang, M. / Caffrey, M. | ||||||
| Funding support |  Ireland, 1items Ireland, 1items
| ||||||
 Citation Citation | Journal: Acta Crystallogr D Struct Biol / Year: 2016 Title: In meso in situ serial X-ray crystallography of soluble and membrane proteins at cryogenic temperatures. Authors: Huang, C.Y. / Olieric, V. / Ma, P. / Howe, N. / Vogeley, L. / Liu, X. / Warshamanage, R. / Weinert, T. / Panepucci, E. / Kobilka, B. / Diederichs, K. / Wang, M. / Caffrey, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5d58.cif.gz | 115.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5d58.ent.gz | 90.1 KB | Display | PDB format |
| PDBx/mmJSON format | 5d58.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/d5/5d58ftp://data.pdbj.org/pub/pdb/validation_reports/d5/5d58 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5d52C  5d53C  5d54C  5d56C  5d57C  5d59C  5d5aC  5d5bC  5d5cC  5d5dC  5d5eC  5d5fC  4d2cS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 52782.148 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus thermophilus (strain ATCC BAA-250 / LMG 18311) (bacteria)Strain: ATCC BAA-250 / LMG 18311 / Gene: dtpT, stu0970 / Production host: |
|---|
-Non-polymers , 6 types, 90 molecules 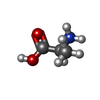



| #2: Chemical | ChemComp-ALA /  Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 Type: L-peptide linking / Mass: 89.093 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2 | ||||
|---|---|---|---|---|---|
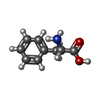
| #3: Chemical | ChemComp-PHE /  Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H11NO2 Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H11NO2 | ||||
| #4: Chemical | ChemComp-PO4 /  Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 | ||||
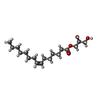
| #5: Chemical | ChemComp-78M / (  Mass: 314.460 Da / Num. of mol.: 21 / Source method: obtained synthetically / Formula: C18H34O4 Mass: 314.460 Da / Num. of mol.: 21 / Source method: obtained synthetically / Formula: C18H34O4#6: Chemical |  Mass: 398.489 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H38O9 / Comment: precipitant*YM Mass: 398.489 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H38O9 / Comment: precipitant*YM#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.88 Å3/Da / Density % sol: 57.33 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: lipidic cubic phase Details: 250-325 mM NH4H2PO4, 100 mM HEPES, pH 7.0, 21-22 %(v/v) PEG 400 and 10 mM Ala-Phe PH range: pH 7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 0.97852 Å / Beamline: X10SA / Wavelength: 0.97852 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Feb 18, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97852 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→50 Å / Num. obs: 24159 / % possible obs: 99.4 % / Redundancy: 4.3 % / Net I/σ(I): 9.31 |
| Reflection shell | Resolution: 2.4→2.46 Å / Redundancy: 4.2 % / Mean I/σ(I) obs: 2.18 / % possible all: 98.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4d2c Resolution: 2.4→45.682 Å / SU ML: 0.31 / Cross valid method: THROUGHOUT / σ(F): 1.35 / Phase error: 26.78 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→45.682 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
