 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6yog: Structure of PepTSt from COC IMISX setup collected by still seria... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6yog | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of PepTSt from COC IMISX setup collected by still serial crystallography on crystals prelocated by 2D X-ray phase-contrast imaging | ||||||
 Components Components | Di-or tripeptide:H+ symporter | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / 2D X-ray phase-contrast imaging / IMISX / in situ rotation images / prelocation / still images / serial crystallography / HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationtripeptide transmembrane transport / tripeptide transmembrane transporter activity / peptide:proton symporter activity / dipeptide transmembrane transporter activity / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Streptococcus thermophilus (bacteria) Streptococcus thermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Huang, C.-Y. / Martiel, I. / Villanueva-Perez, P. / Panepucci, E. / Caffrey, M. / Wang, M. | ||||||
 Citation Citation | Journal: Iucrj / Year: 2020 Title: Low-dose in situ prelocation of protein microcrystals by 2D X-ray phase-contrast imaging for serial crystallography. Authors: Martiel, I. / Huang, C.Y. / Villanueva-Perez, P. / Panepucci, E. / Basu, S. / Caffrey, M. / Pedrini, B. / Bunk, O. / Stampanoni, M. / Wang, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6yog.cif.gz | 113.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6yog.ent.gz | 88.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6yog.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yo/6yogftp://data.pdbj.org/pub/pdb/validation_reports/yo/6yog | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6yobC  6yocC  6yodC  6yoeC  6yofC  5d58S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 52782.148 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus thermophilus (strain ATCC BAA-250 / LMG 18311) (bacteria)Gene: dtpT, stu0970 / Production host: |
|---|
-Non-polymers , 5 types, 100 molecules 


| #2: Chemical | ChemComp-PO4 /  Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 | ||||||
|---|---|---|---|---|---|---|---|
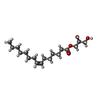
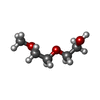
| #3: Chemical | ChemComp-78M / (  Mass: 314.460 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C18H34O4 Mass: 314.460 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C18H34O4#4: Chemical | ChemComp-PG0 / |  Mass: 120.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H12O3 / Comment: precipitant*YM Mass: 120.147 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H12O3 / Comment: precipitant*YM#5: Chemical | ChemComp-PGE / |  Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O4#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 77 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.99 Å3/Da / Density % sol: 58.84 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: lipidic cubic phase / pH: 7 Details: 250-325 mM NH4H2PO4, 100 mM HEPES, pH 7.0, 21-22 %(v/v) PEG 400 and 10 mM Ala-Phe |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: Y |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Dec 28, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→55.64 Å / Num. obs: 28437 / % possible obs: 100 % / Redundancy: 51.9 % / CC1/2: 0.92 / Rrim(I) all: 0.17 / Net I/σ(I): 3.73 |
| Reflection shell | Resolution: 2.3→2.38 Å / Redundancy: 32.6 % / Num. unique obs: 2806 / CC1/2: 0.33 / Rrim(I) all: 2.15 / % possible all: 99.95 |
| Serial crystallography sample delivery | Method: fixed target |
| Serial crystallography sample delivery fixed target | Description: IMISX / Sample dehydration prevention: COC sandwich / Sample holding: IMISX / Support base: 3D-printed holder in standard goniometer base |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5D58 Resolution: 2.3→55.64 Å / SU ML: 0.3 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 28.03
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 122.69 Å2 / Biso mean: 58.2949 Å2 / Biso min: 27.05 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.3→55.64 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / % reflection obs: 100 %
|
