National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI)
P01AI104722
United States
Citation
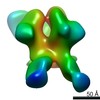
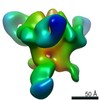
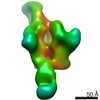
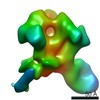
Journal: Immunity / Year: 2019 Title: Vaccination with Glycan-Modified HIV NFL Envelope Trimer-Liposomes Elicits Broadly Neutralizing Antibodies to Multiple Sites of Vulnerability. Authors: Viktoriya Dubrovskaya / Karen Tran / Gabriel Ozorowski / Javier Guenaga / Richard Wilson / Shridhar Bale / Christopher A Cottrell / Hannah L Turner / Gemma Seabright / Sijy O'Dell / Jonathan ...Authors: Viktoriya Dubrovskaya / Karen Tran / Gabriel Ozorowski / Javier Guenaga / Richard Wilson / Shridhar Bale / Christopher A Cottrell / Hannah L Turner / Gemma Seabright / Sijy O'Dell / Jonathan L Torres / Lifei Yang / Yu Feng / Daniel P Leaman / Néstor Vázquez Bernat / Tyler Liban / Mark Louder / Krisha McKee / Robert T Bailer / Arlette Movsesyan / Nicole A Doria-Rose / Marie Pancera / Gunilla B Karlsson Hedestam / Michael B Zwick / Max Crispin / John R Mascola / Andrew B Ward / Richard T Wyatt / Abstract: The elicitation of broadly neutralizing antibodies (bNAbs) against the HIV-1 envelope glycoprotein (Env) trimer remains a major vaccine challenge. Most cross-conserved protein determinants are ...The elicitation of broadly neutralizing antibodies (bNAbs) against the HIV-1 envelope glycoprotein (Env) trimer remains a major vaccine challenge. Most cross-conserved protein determinants are occluded by self-N-glycan shielding, limiting B cell recognition of the underlying polypeptide surface. The exceptions to the contiguous glycan shield include the conserved receptor CD4 binding site (CD4bs) and glycoprotein (gp)41 elements proximal to the furin cleavage site. Accordingly, we performed heterologous trimer-liposome prime:boosting in rabbits to drive B cells specific for cross-conserved sites. To preferentially expose the CD4bs to B cells, we eliminated proximal N-glycans while maintaining the native-like state of the cleavage-independent NFL trimers, followed by gradual N-glycan restoration coupled with heterologous boosting. This approach successfully elicited CD4bs-directed, cross-neutralizing Abs, including one targeting a unique glycan-protein epitope and a bNAb (87% breadth) directed to the gp120:gp41 interface, both resolved by high-resolution cryoelectron microscopy. This study provides proof-of-principle immunogenicity toward eliciting bNAbs by vaccination.
Mass: 25017.061 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host: Homo sapiens (human)
#2: Antibody
1C2FabLightChain
Mass: 22830.307 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host: Homo sapiens (human)
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation

 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
























 PDBj
PDBj


 Assembly
Assembly



 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 268 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing