 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4yxs: CAMP-DEPENDENT PROTEIN KINASE PKA CATALYTIC SUBUNIT WITH PKI-5-24 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4yxs | ||||||
|---|---|---|---|---|---|---|---|
| Title | CAMP-DEPENDENT PROTEIN KINASE PKA CATALYTIC SUBUNIT WITH PKI-5-24 | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE/INHIBITOR / ATP-BINDING / PHOSPHOPROTEIN / SERINE/THREONINE-PROTEIN KINASE / TRANSFERASE / CAMP / PROTEIN KINASE INHIBITOR / TRANSFERASE-IN COMPLEX / TRANSFERASE-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationCD209 (DC-SIGN) signaling / HDL assembly / Regulation of insulin secretion / Rap1 signalling / Ion homeostasis / PKA activation in glucagon signalling / DARPP-32 events / CREB1 phosphorylation through the activation of Adenylate Cyclase / GPER1 signaling / Loss of Nlp from mitotic centrosomes ...CD209 (DC-SIGN) signaling / HDL assembly / Regulation of insulin secretion / Rap1 signalling / Ion homeostasis / PKA activation in glucagon signalling / DARPP-32 events / CREB1 phosphorylation through the activation of Adenylate Cyclase / GPER1 signaling / Loss of Nlp from mitotic centrosomes / Recruitment of mitotic centrosome proteins and complexes / Loss of proteins required for interphase microtubule organization from the centrosome / Anchoring of the basal body to the plasma membrane / AURKA Activation by TPX2 / Factors involved in megakaryocyte development and platelet production / RET signaling / Interleukin-3, Interleukin-5 and GM-CSF signaling / negative regulation of cAMP-dependent protein kinase activity / Recruitment of NuMA to mitotic centrosomes / VEGFA-VEGFR2 Pathway / PKA activation / MAPK6/MAPK4 signaling / GLI3 is processed to GLI3R by the proteasome / Regulation of PLK1 Activity at G2/M Transition / Hedgehog 'off' state / negative regulation of cAMP/PKA signal transduction / cAMP-dependent protein kinase inhibitor activity / cAMP-dependent protein kinase / regulation of protein processing / cAMP-dependent protein kinase activity / protein localization to lipid droplet / cAMP-dependent protein kinase complex / regulation of bicellular tight junction assembly / cellular response to parathyroid hormone stimulus / molecular function inhibitor activity / negative regulation of protein import into nucleus / cellular response to cold / Glucagon-like Peptide-1 (GLP1) regulates insulin secretion / regulation of osteoblast differentiation / sperm capacitation / Mitochondrial protein degradation / High laminar flow shear stress activates signaling by PIEZO1 and PECAM1:CDH5:KDR in endothelial cells / Vasopressin regulates renal water homeostasis via Aquaporins / ciliary base / negative regulation of glycolytic process through fructose-6-phosphate / protein kinase A regulatory subunit binding / protein kinase A catalytic subunit binding / mesoderm formation / sperm flagellum / plasma membrane raft / axoneme / postsynaptic modulation of chemical synaptic transmission / regulation of G2/M transition of mitotic cell cycle / negative regulation of TORC1 signaling / regulation of proteasomal protein catabolic process / positive regulation of gluconeogenesis / protein serine/threonine/tyrosine kinase activity / cellular response to glucagon stimulus / protein export from nucleus / acrosomal vesicle / positive regulation of protein export from nucleus / negative regulation of smoothened signaling pathway / neural tube closure / positive regulation of cholesterol biosynthetic process / cellular response to glucose stimulus / adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway / neuromuscular junction / positive regulation of insulin secretion / adenylate cyclase-activating G protein-coupled receptor signaling pathway / mRNA processing / peptidyl-serine phosphorylation / manganese ion binding / cellular response to heat / molecular adaptor activity / postsynapse / protein kinase activity / regulation of cell cycle / nuclear speck / protein phosphorylation / protein domain specific binding / protein serine kinase activity / protein serine/threonine kinase activity / centrosome / ubiquitin protein ligase binding / protein kinase binding / perinuclear region of cytoplasm / glutamatergic synapse / negative regulation of transcription by RNA polymerase II / magnesium ion binding / signal transduction / mitochondrion / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.11 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 2.11 Å | ||||||
 Authors Authors | Schiffer, A. / Wendt, K.U. | ||||||
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2015 Title: A combination of spin diffusion methods for the determination of protein-ligand complex structural ensembles. Authors: Pilger, J. / Mazur, A. / Monecke, P. / Schreuder, H. / Elshorst, B. / Bartoschek, S. / Langer, T. / Schiffer, A. / Krimm, I. / Wegstroth, M. / Lee, D. / Hessler, G. / Wendt, K.U. / Becker, S. / Griesinger, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4yxs.cif.gz | 96.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4yxs.ent.gz | 71.6 KB | Display | PDB format |
| PDBx/mmJSON format | 4yxs.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/yx/4yxsftp://data.pdbj.org/pub/pdb/validation_reports/yx/4yxs | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4c4xC  4c4yC  4c4zC  4yxrC  4exf C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 40707.402 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Protein/peptide | Mass: 2226.411 Da / Num. of mol.: 1 / Fragment: UNP RESIDUES 6-25 / Source method: obtained synthetically / Source: (synth.) |
| #3: Chemical | ChemComp-EMU / 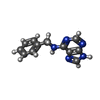  Mass: 225.249 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H11N5 / Comment: inhibitor*YM Mass: 225.249 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H11N5 / Comment: inhibitor*YM |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 343 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 343 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.57 Å3/Da / Density % sol: 52.1 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 6.4 Details: 75MM LICL, 30MM MESBISTRIS, 1MM DTT, 0.2MM EDTA, PH 6.5, 15% METHANOL, PH 6.4, VAPOR DIFFUSION, HANGING DROP, TEMPERATURE 277K PH range: 6.4 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 1 Å / Beamline: ID23-1 / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Sep 9, 2010 |
| Radiation | Monochromator: SILICON (1 1 1) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.11→75.35 Å / Num. obs: 26146 / % possible obs: 99.9 % / Observed criterion σ(I): 1 / Redundancy: 5.5 % / Biso Wilson estimate: 25.96 Å2 / Net I/σ(I): 15 |
| Reflection shell | Resolution: 2.11→2.22 Å / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 2.11→53.99 Å / Cor.coef. Fo:Fc: 0.9348 / Cor.coef. Fo:Fc free: 0.8868 / SU R Cruickshank DPI: 0.19 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.209 / SU Rfree Blow DPI: 0.18 / SU Rfree Cruickshank DPI: 0.174
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.11 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.223 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.11→53.99 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.11→2.2 Å / Total num. of bins used: 13
|
