Entry Database : PDB / ID : 4ogbTitle Crystal structure of the catalytic domain of PDE4D2 with compound 2 cAMP-specific 3',5'-cyclic phosphodiesterase 4D Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / / Resolution : 2.032 Å Authors Feil, S.C. / Parker, M.W. Journal : To be Published Title : The PDE inhibition profile of LY294002 and tetrahydropyranyl analogues reveals a chromone motif for the development of PDE inhibitorsAuthors : Howard, B.L. / Nankervis, J.L. / Feil, S.C. / Manallack, D.T. / Holien, J.K. / Zhen, Z. / Jennings, I.G. / Abbott, B.M. / Parker, M.W. / Thompson, P.E. History Deposition Jan 15, 2014 Deposition site / Processing site Revision 1.0 Jan 21, 2015 Provider / Type Revision 1.1 Nov 22, 2017 Group / Category / Item Revision 1.2 Feb 28, 2024 Group / Database references / Derived calculationsCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_struct_conn_angle / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_asym_id / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_asym_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






















 PDBj
PDBj



 Assembly
Assembly





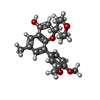






 Mass: 382.450 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C23H26O5
Mass: 382.450 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C23H26O5 Mass: 62.068 Da / Num. of mol.: 32 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 32 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn Mass: 78.133 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM
Mass: 78.133 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM
Mass: 238.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Sample preparation
Sample preparation / Beamline: MX1 / Wavelength: 0.95 Å
/ Beamline: MX1 / Wavelength: 0.95 Å Processing
Processing