Entry Database : PDB / ID : 1y2dTitle Catalytic Domain Of Human Phosphodiesterase 4D In Complex With 1-(4-methoxy-phenyl)-3,5-dimethyl-1H-pyrazole-4-carboxylic acid ethyl ester cAMP-specific 3',5'-cyclic phosphodiesterase 4D Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 1.7 Å Authors Card, G.L. / Blasdel, L. / England, B.P. / Zhang, C. / Suzuki, Y. / Gillette, S. / Fong, D. / Ibrahim, P.N. / Artis, D.R. / Bollag, G. ...Card, G.L. / Blasdel, L. / England, B.P. / Zhang, C. / Suzuki, Y. / Gillette, S. / Fong, D. / Ibrahim, P.N. / Artis, D.R. / Bollag, G. / Milburn, M.V. / Kim, S.-H. / Schlessinger, J. / Zhang, K.Y.J. Journal : Nat.Biotechnol. / Year : 2005Title : A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug designAuthors : Card, G.L. / Blasdel, L. / England, B.P. / Zhang, C. / Suzuki, Y. / Gillette, S. / Fong, D. / Ibrahim, P.N. / Artis, D.R. / Bollag, G. / Milburn, M.V. / Kim, S.-H. / Schlessinger, J. / Zhang, K.Y.J. History Deposition Nov 22, 2004 Deposition site / Processing site Revision 1.0 Mar 1, 2005 Provider / Type Revision 1.1 Apr 30, 2008 Group Revision 1.2 Jul 13, 2011 Group / Version format complianceRevision 1.3 Feb 14, 2024 Group Advisory / Data collection ... Advisory / Data collection / Database references / Derived calculations / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_database_remark / pdbx_struct_conn_angle / struct_conn / struct_ncs_dom_lim / struct_ref_seq_dif / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_database_remark.text / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_asym_id / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_asym_id / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_ncs_dom_lim.beg_auth_comp_id / _struct_ncs_dom_lim.end_auth_comp_id / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less Remark 600 HETEROGEN HOH 1003-1008 ARE ASSOCIATED WITH CHAIN A. HOH 2003-2008 ARE ASSOCIATED WITH CHAIN B.
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

























 PDBj
PDBj



 Assembly
Assembly





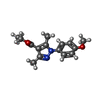



 Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 274.315 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H18N2O3
Mass: 274.315 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H18N2O3 Mass: 62.068 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 282.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H26N2O6 / Comment: pH buffer*YM
Mass: 282.334 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H26N2O6 / Comment: pH buffer*YM Sample preparation
Sample preparation / Beamline: 8.3.1 / Wavelength: 1.1 Å
/ Beamline: 8.3.1 / Wavelength: 1.1 Å Processing
Processing