 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4dxl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of IspE (4-diphosphocytidyl-2-C-methyl-D-erythritol kinase) from Mycobacterium abscessus, bound to CMP and ATP | ||||||
 Components Components | 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase | ||||||
 Keywords Keywords | TRANSFERASE / SSGCID / NIH / NIAID / SBRI / Emerald BioStructures / Structural Genomics / Seattle Structural Genomics Center for Infectious Disease | ||||||
| Function / homology |  Function and homology information Function and homology information4-(cytidine 5'-diphospho)-2-C-methyl-D-erythritol kinase / 4-(cytidine 5'-diphospho)-2-C-methyl-D-erythritol kinase activity / isopentenyl diphosphate biosynthetic process, methylerythritol 4-phosphate pathway / terpenoid biosynthetic process / ATP binding Similarity search - Function | ||||||
| Biological species |  Mycobacterium abscessus (bacteria) Mycobacterium abscessus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Seattle Structural Genomics Center for Infectious Disease (SSGCID) | ||||||
 Citation Citation | Journal: Tuberculosis (Edinb) / Year: 2015 Title: Increasing the structural coverage of tuberculosis drug targets. Authors: Baugh, L. / Phan, I. / Begley, D.W. / Clifton, M.C. / Armour, B. / Dranow, D.M. / Taylor, B.M. / Muruthi, M.M. / Abendroth, J. / Fairman, J.W. / Fox, D. / Dieterich, S.H. / Staker, B.L. / ...Authors: Baugh, L. / Phan, I. / Begley, D.W. / Clifton, M.C. / Armour, B. / Dranow, D.M. / Taylor, B.M. / Muruthi, M.M. / Abendroth, J. / Fairman, J.W. / Fox, D. / Dieterich, S.H. / Staker, B.L. / Gardberg, A.S. / Choi, R. / Hewitt, S.N. / Napuli, A.J. / Myers, J. / Barrett, L.K. / Zhang, Y. / Ferrell, M. / Mundt, E. / Thompkins, K. / Tran, N. / Lyons-Abbott, S. / Abramov, A. / Sekar, A. / Serbzhinskiy, D. / Lorimer, D. / Buchko, G.W. / Stacy, R. / Stewart, L.J. / Edwards, T.E. / Van Voorhis, W.C. / Myler, P.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4dxl.cif.gz | 129.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4dxl.ent.gz | 98.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4dxl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dx/4dxlftp://data.pdbj.org/pub/pdb/validation_reports/dx/4dxl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3gvcC  3gvgC  3gwcC  3h7fC  3h81C  3he2C  3hwiC  3hwkC  3hzgC  3icoC  3khpC  3llsC  3moyC  3mpzC  3mybC  3ndnC  3ndoC  3nf4C  3ng3C  3njdC  3nwoC  3o0mC  3o38C  3oc6C  3oc7C  3oi9C  3oksC  3omeC  3p0tC  3p2yC  3p4iC  3p4tC  3p5mC  3p85C  3pe8C  3pk0C  3ppiC  3pzyC  3q1tC  3q8nC  3qbpC  3qdfC  3qhaC  3qivC  3qk8C  3qkaC  3qljC  3qmjC  3qreC  3quaC  3quvC  3qxiC  3qxzC  3qyrC  3r0oC  3r1iC  3r1jC  3r20C  3r2nC  3r4tC  3r6hC  3r6oC  3r7kC  3r8cC  3r9pC  3r9qC  3r9rC  3r9sC  3r9tC  3rd5C  3rd7C  3rd8C  3rfqC  3rihC  3rr2C  3rr6C  3rrpC  3rrvC  3rsiC  3rv2C  3s82C  3sbxC  3sf6C  3sllC  3svkC  3svtC  3swoC  3swtC  3swxC  3t3wC  3tavC  3tcrC  3tdeC  3tjrC  3tl3C  3tlfC  3trrC  3tx2C  3tzqC  3tzuC  3u0aC  3ucxC  3uveC  4di1C  4dieC  4dq8C  4ed4C  4egeC  4egfC  4emdC 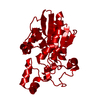 4eo9C  4eyeC  4f3wC  4f47C  4ffcC  4gk6C  4hdtC  4hr3C  4i1yC  4ijnC  4iv6C  4iz9C  4j5iC  4kamC  4lgvC  4o2dC  3pygS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 32742.580 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Mycobacterium abscessus (bacteria) / Strain: ATCC 19977 / DSM 44196 / Gene: ispE, MAB_1139 / Plasmid: AVA0421 / Production host: References: UniProt: B1MKD5, 4-(cytidine 5'-diphospho)-2-C-methyl-D-erythritol kinase |
|---|---|
| #2: Chemical | ChemComp-C5P / 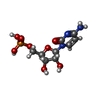  Mass: 323.197 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N3O8P Mass: 323.197 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H14N3O8P |
| #3: Chemical | ChemComp-ATP /   Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM |
| #4: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 215 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 215 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.09 Å3/Da / Density % sol: 41.06 % |
|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, sitting drop / pH: 4.5 Details: Internal tracking number 230819a2, INDEX screen condition A2: 2 M ammonium sulfate, 0.1 M acetate, pH 4.5, MyabA.00725.a.A1 PW30213 at 29.3 mg/mL in 25 mM HEPES, pH 7.0, 500 mM sodium ...Details: Internal tracking number 230819a2, INDEX screen condition A2: 2 M ammonium sulfate, 0.1 M acetate, pH 4.5, MyabA.00725.a.A1 PW30213 at 29.3 mg/mL in 25 mM HEPES, pH 7.0, 500 mM sodium chloride, 2 mM DTT, 0.025% sodium azide, 5% glycerol, 2 mM CDP, 2 mM ATP, 2 mM meso-erythritol, cryoprotectant: Al's oil, VAPOR DIFFUSION, SITTING DROP, temperature 290K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E SUPERBRIGHT / Wavelength: 1.54178 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: RIGAKU SATURN 944+ / Detector: CCD / Date: Feb 23, 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54178 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→50 Å / Num. obs: 17719 / % possible obs: 92.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 5.3 % / Biso Wilson estimate: 18.285 Å2 / Rmerge(I) obs: 0.073 / Net I/σ(I): 16.65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3PYG Resolution: 2→35.562 Å / Cor.coef. Fo:Fc: 0.916 / Cor.coef. Fo:Fc free: 0.838 / SU B: 7.914 / SU ML: 0.126 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.241 / ESU R Free: 0.203 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: U VALUES : WITH TLS ADDED HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 12.101 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→35.562 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.052 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
