 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2bhd | ||||||
|---|---|---|---|---|---|---|---|
| Title | Mg substituted E. coli Aminopeptidase P in complex with product | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / HYDROLASE-SUBSTRATE COMPLEX / PROLINE-SPECIFIC PEPTIDASE / PRODUCT COMPLEX / METALLOENZYME / PITA-BREAD FOLD / DINUCLEAR HYDROLASE / AMINOPEPTIDASE / MANGANESE / METAL-BINDING / METALLOPROTEASE | ||||||
| Function / homology |  Function and homology information Function and homology informationXaa-Pro aminopeptidase / metalloexopeptidase activity / metalloaminopeptidase activity / aminopeptidase activity / manganese ion binding / protein homotetramerization / protein-containing complex / proteolysis / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  SYNTHETIC CONSTRUCT (others) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Graham, S.C. / Bond, C.S. / Freeman, H.C. / Guss, J.M. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2005 Title: Structural and Functional Implications of Metal Ion Selection in Aminopeptidase P, a Metalloprotease with a Dinuclear Metal Center. Authors: Graham, S.C. / Bond, C.S. / Freeman, H.C. / Guss, J.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2bhd.cif.gz | 103.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2bhd.ent.gz | 78.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2bhd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bh/2bhdftp://data.pdbj.org/pub/pdb/validation_reports/bh/2bhd | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1w2mC  1w7vC  1wbqC  1wl6C  1wl9C  1wlrC  2bh3C  2bhaC  2bhbC  2bhcC  2bn7C  1n51S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |





























-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | AMINOPEPTIDASE P IS A HOMOTETRAMER IN SOLUTION. HOWEVER, SINCE IN THIS ENTRY, EACH CHAIN OF THIS PROTEIN IS IN COMPLEX WITH A TRIPEPTIDE, THIS ENTRY IS CLASSIFIED AS A HETEROOCTAMER. |
-Components
| #1: Protein | Mass: 49744.062 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) | ||||
|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 327.419 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: TRIPEPTIDE SUBSTRATE OF AMINOPEPTIDASE P / Source: (synth.) SYNTHETIC CONSTRUCT (others) | ||||
| #3: Chemical |   Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg#4: Chemical | ChemComp-FLC / | 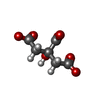  Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 98 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 5.6 Å3/Da / Density % sol: 77.8 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: AMINOPEPTIDASE P WAS DIALYSED AGAINST EGTA PRIOR TO CRYSTALLISATION. CRYSTALS WERE GROWN USING HANGING DROP VAOPR DIFFUSION AT 4C. RESERVOIR CONTAINED 26% MPD, 100 MM NA.CITRATE (PH 7.5) AND ...Details: AMINOPEPTIDASE P WAS DIALYSED AGAINST EGTA PRIOR TO CRYSTALLISATION. CRYSTALS WERE GROWN USING HANGING DROP VAOPR DIFFUSION AT 4C. RESERVOIR CONTAINED 26% MPD, 100 MM NA.CITRATE (PH 7.5) AND 200 MM MG.ACETATE. CRYSTALS WERE SOAKED IN RESERVOIR SOLUTION SUPPLEMENTED WITH 10 MM VALPROLEU TRIPEPTIDE FOR 20 HOURS AT 4C PRIOR TO DATA COLLECTION. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200H / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Oct 28, 2004 / Details: OSMIC MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→60 Å / Num. obs: 38892 / % possible obs: 99.2 % / Observed criterion σ(I): -3 / Redundancy: 7.8 % / Biso Wilson estimate: 55.48 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 22.7 |
| Reflection shell | Resolution: 2.5→2.59 Å / Redundancy: 7 % / Rmerge(I) obs: 0.58 / Mean I/σ(I) obs: 3.4 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1N51 STRIPPED OF MULTIPLE CONFORMERS, SOLVENT ATOMS AND HETERO COMPOUNDS Resolution: 2.5→60.19 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.946 / SU ML: 0.106 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.184 / ESU R Free: 0.163 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. SUFFICIENT DENSITY WAS NOT PRESENT TO ALLOW FULL MODELLING OF RESIDUES A439 AND A440. A PROLEU DIPEPTIDE IS VISIBLE IN THE ACTIVE SITE. IT ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. SUFFICIENT DENSITY WAS NOT PRESENT TO ALLOW FULL MODELLING OF RESIDUES A439 AND A440. A PROLEU DIPEPTIDE IS VISIBLE IN THE ACTIVE SITE. IT IS ASSUMED THAT CONTAMINATING ACTIVITY IN THE DROP CLEAVED ALL OF THE VALPROLEU PEPTIDE PRESENT IN THE DROP, LEAVING ONLY PROLEU WHICH BINDS TO THE ENZYME AS A PRODUCT INHIBITOR.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 43.16 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→60.19 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|